Lapek John D, Greninger Patricia, Morris Robert, Amzallag Arnaud, Pruteanu-Malinici Iulian, Benes Cyril H, Haas Wilhelm
Massachusetts General Hospital Cancer Center and Department of Medicine, Harvard Medical School, Charlestown, Massachusetts, USA.
Nat Biotechnol. 2017 Oct;35(10):983-989. doi: 10.1038/nbt.3955. Epub 2017 Sep 11.
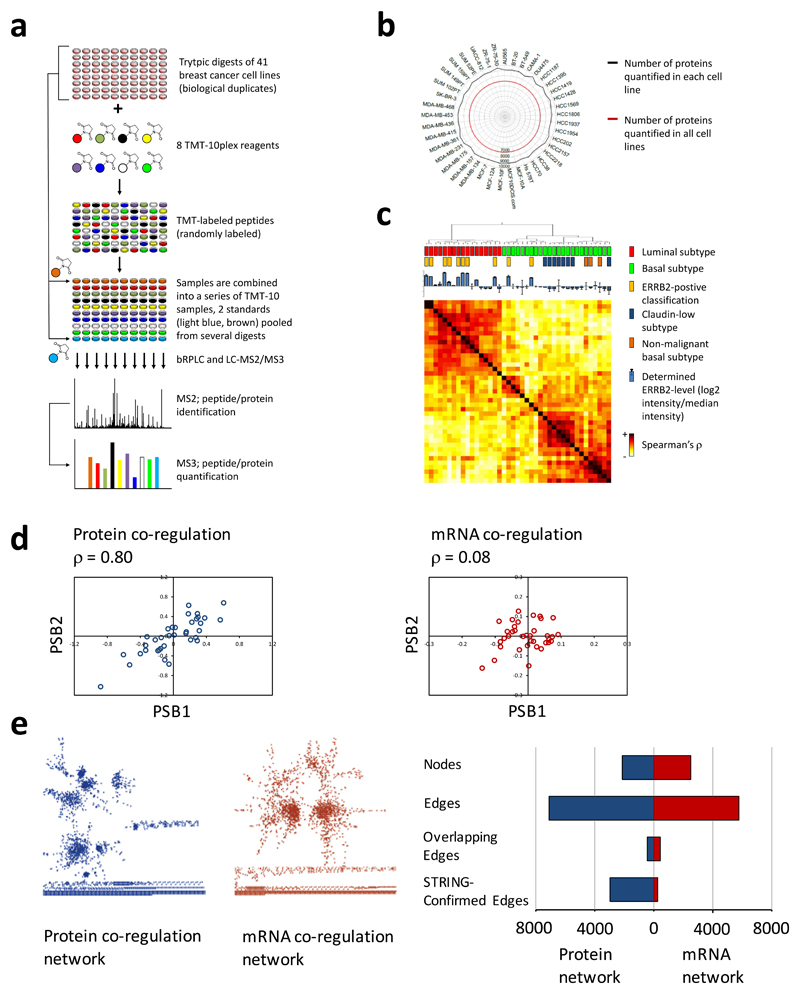
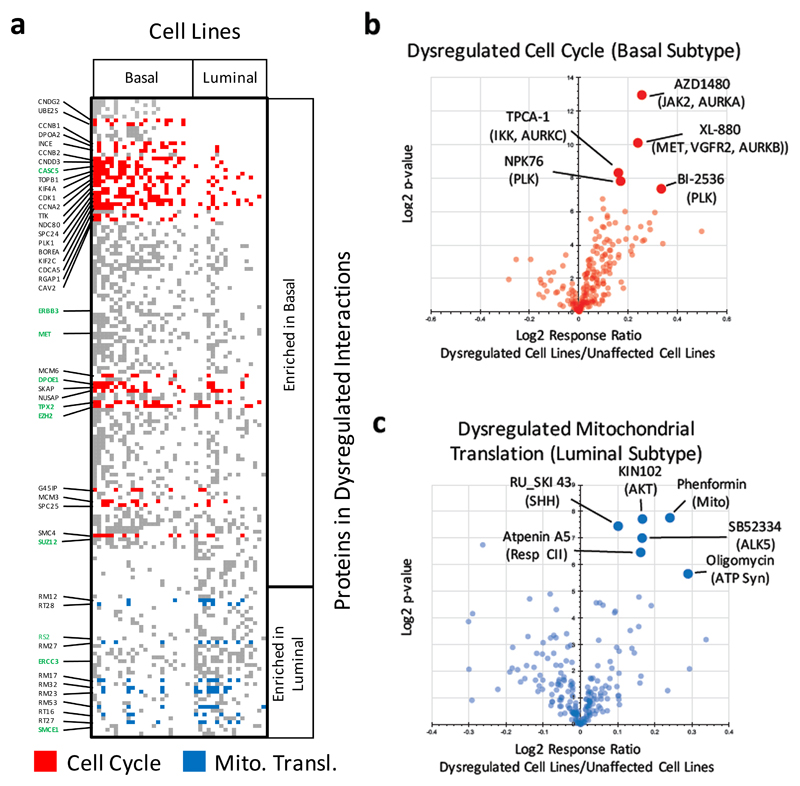
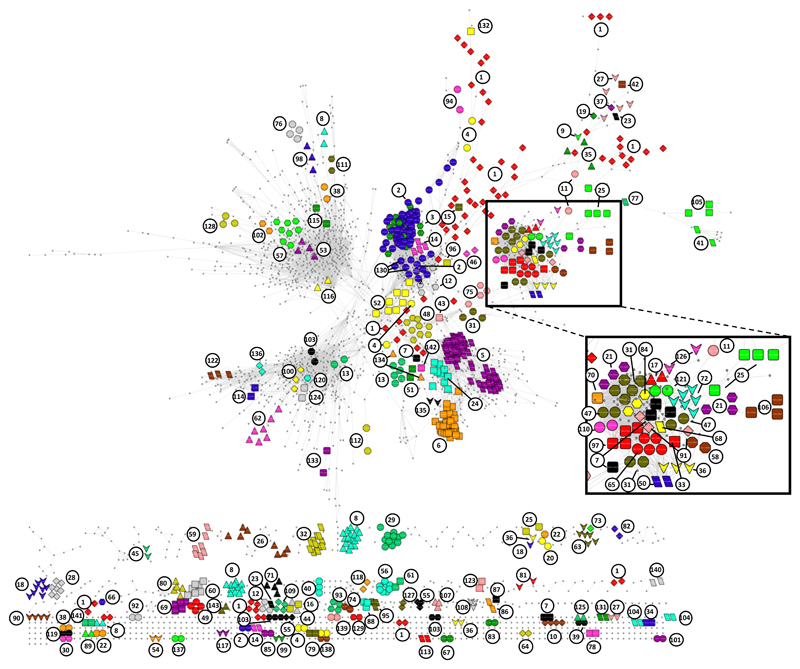
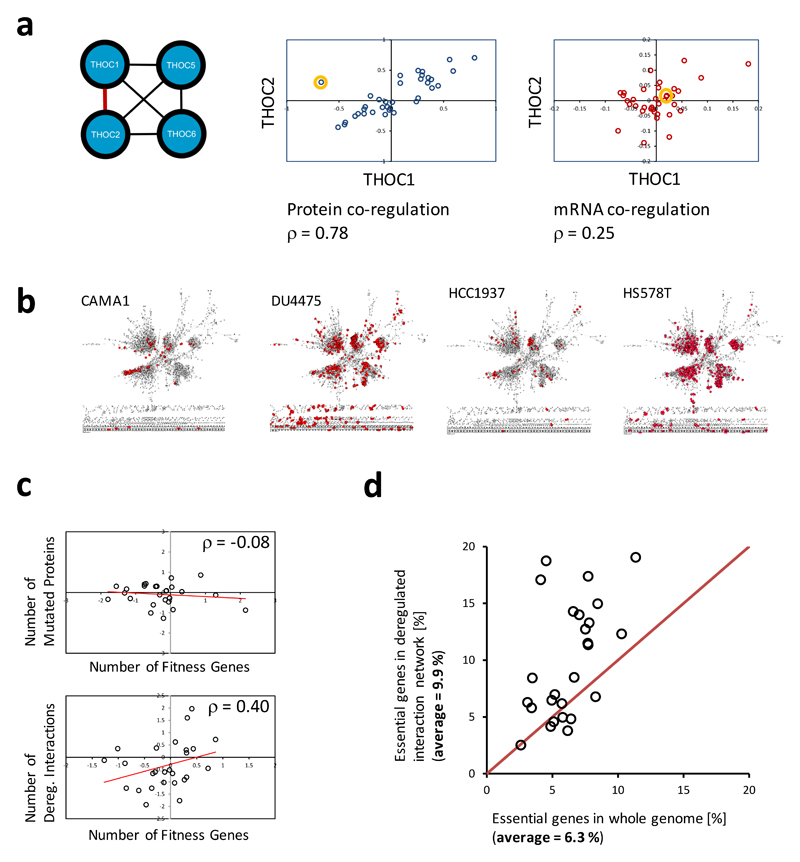
The formation of protein complexes and the co-regulation of the cellular concentrations of proteins are essential mechanisms for cellular signaling and for maintaining homeostasis. Here we use isobaric-labeling multiplexed proteomics to analyze protein co-regulation and show that this allows the identification of protein-protein associations with high accuracy. We apply this 'interactome mapping by high-throughput quantitative proteome analysis' (IMAHP) method to a panel of 41 breast cancer cell lines and show that deviations of the observed protein co-regulations in specific cell lines from the consensus network affects cellular fitness. Furthermore, these aberrant interactions serve as biomarkers that predict the drug sensitivity of cell lines in screens across 195 drugs. We expect that IMAHP can be broadly used to gain insight into how changing landscapes of protein-protein associations affect the phenotype of biological systems.
蛋白质复合物的形成以及蛋白质细胞浓度的协同调节是细胞信号传导和维持体内平衡的重要机制。在此,我们使用等压标记多重蛋白质组学来分析蛋白质的协同调节,并表明这能够高精度地鉴定蛋白质-蛋白质相互作用。我们将这种“通过高通量定量蛋白质组分析进行相互作用组图谱绘制”(IMAHP)方法应用于一组41种乳腺癌细胞系,结果表明特定细胞系中观察到的蛋白质协同调节与共识网络的偏差会影响细胞适应性。此外,这些异常相互作用可作为生物标志物,预测195种药物筛选中细胞系的药物敏感性。我们期望IMAHP能够广泛用于深入了解蛋白质-蛋白质相互作用格局的变化如何影响生物系统的表型。