Zhou Jia, Sears Renee L, Xing Xiaoyun, Zhang Bo, Li Daofeng, Rockweiler Nicole B, Jang Hyo Sik, Choudhary Mayank N K, Lee Hyung Joo, Lowdon Rebecca F, Arand Jason, Tabers Brianne, Gu C Charles, Cicero Theodore J, Wang Ting
Department of Genetics, Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, MO, USA.
Division of Biostatistics, Washington University School of Medicine, St. Louis, MO, USA.
BMC Genomics. 2017 Sep 12;18(1):724. doi: 10.1186/s12864-017-4115-6.
Uncovering mechanisms of epigenome evolution is an essential step towards understanding the evolution of different cellular phenotypes. While studies have confirmed DNA methylation as a conserved epigenetic mechanism in mammalian development, little is known about the conservation of tissue-specific genome-wide DNA methylation patterns.
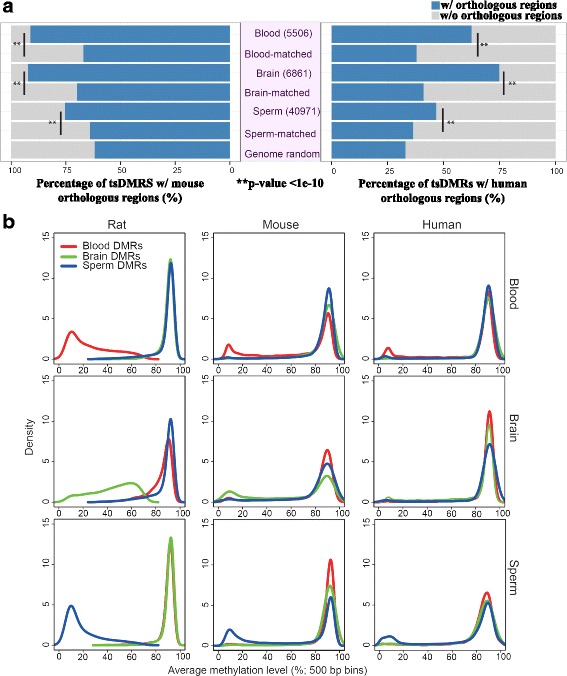
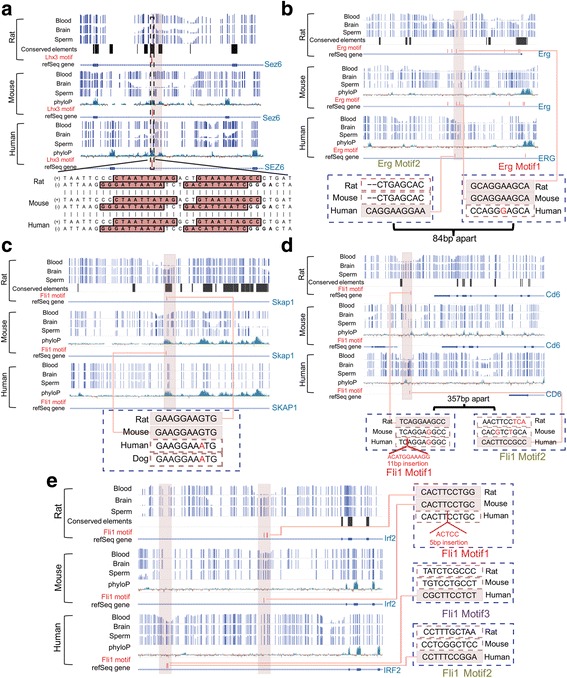
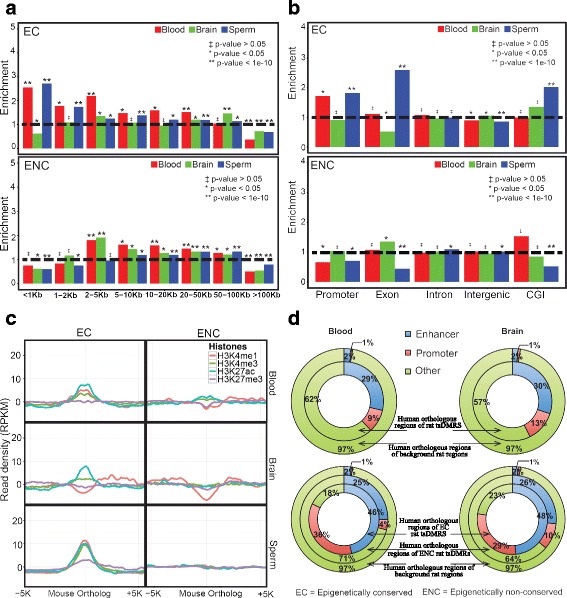
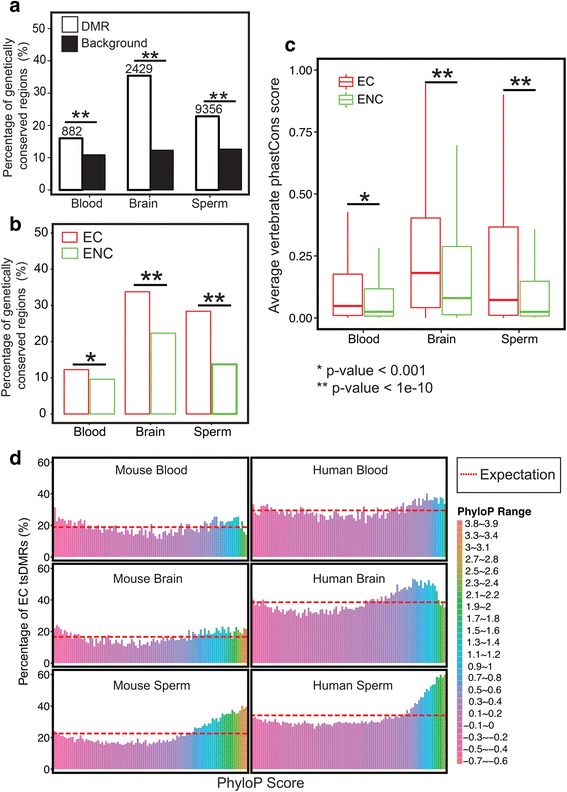
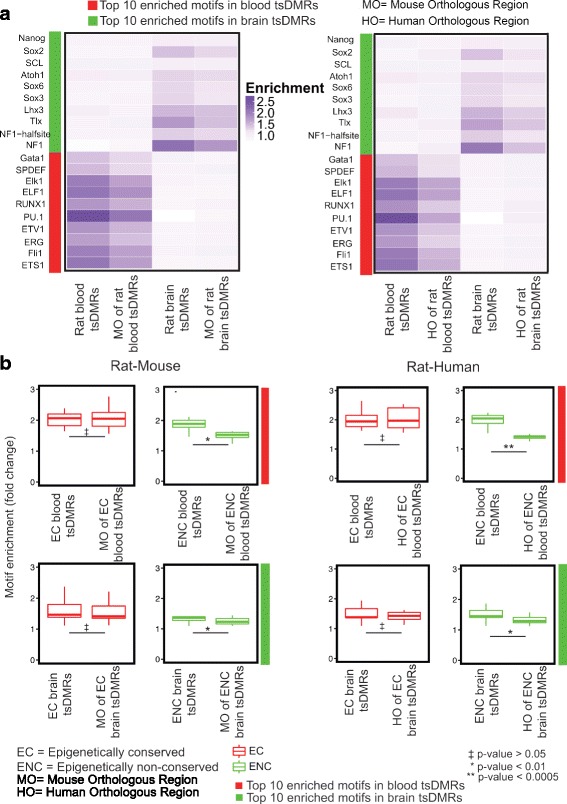
Using a comparative epigenomics approach, we identified and compared the tissue-specific DNA methylation patterns of rat against those of mouse and human across three shared tissue types. We confirmed that tissue-specific differentially methylated regions are strongly associated with tissue-specific regulatory elements. Comparisons between species revealed that at a minimum 11-37% of tissue-specific DNA methylation patterns are conserved, a phenomenon that we define as epigenetic conservation. Conserved DNA methylation is accompanied by conservation of other epigenetic marks including histone modifications. Although a significant amount of locus-specific methylation is epigenetically conserved, the majority of tissue-specific DNA methylation is not conserved across the species and tissue types that we investigated. Examination of the genetic underpinning of epigenetic conservation suggests that primary sequence conservation is a driving force behind epigenetic conservation. In contrast, evolutionary dynamics of tissue-specific DNA methylation are best explained by the maintenance or turnover of binding sites for important transcription factors.
Our study extends the limited literature of comparative epigenomics and suggests a new paradigm for epigenetic conservation without genetic conservation through analysis of transcription factor binding sites.
揭示表观基因组进化机制是理解不同细胞表型进化的关键一步。虽然已有研究证实DNA甲基化是哺乳动物发育过程中一种保守的表观遗传机制,但对于全基因组范围内组织特异性DNA甲基化模式的保守性却知之甚少。
我们采用比较表观基因组学方法,在三种共享组织类型中鉴定并比较了大鼠与小鼠和人类的组织特异性DNA甲基化模式。我们证实,组织特异性差异甲基化区域与组织特异性调控元件密切相关。物种间比较显示,至少11%-37%的组织特异性DNA甲基化模式是保守的,我们将这一现象定义为表观遗传保守性。保守的DNA甲基化伴随着包括组蛋白修饰在内的其他表观遗传标记的保守性。虽然大量位点特异性甲基化在表观遗传上是保守的,但在我们研究的物种和组织类型中,大多数组织特异性DNA甲基化并不保守。对表观遗传保守性的遗传基础研究表明,一级序列保守性是表观遗传保守性背后的驱动力。相比之下,组织特异性DNA甲基化的进化动力学最好通过重要转录因子结合位点的维持或更替来解释。
我们的研究扩展了有限的比较表观基因组学文献,并通过对转录因子结合位点的分析,提出了一种没有遗传保守性的表观遗传保守性新范式。