European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, UK.
Department of Medical Biochemistry and Biophysics, Division of Functional Genomics and Systems Biology, Karolinska Institutet, Stockholm, SE, 141 83, Sweden.
Genome Biol. 2024 Jun 6;25(1):146. doi: 10.1186/s13059-024-03218-6.
DNA methylation is an important epigenetic modification which has numerous roles in modulating genome function. Its levels are spatially correlated across the genome, typically high in repressed regions but low in transcription factor (TF) binding sites and active regulatory regions. However, the mechanisms establishing genome-wide and TF binding site methylation patterns are still unclear.
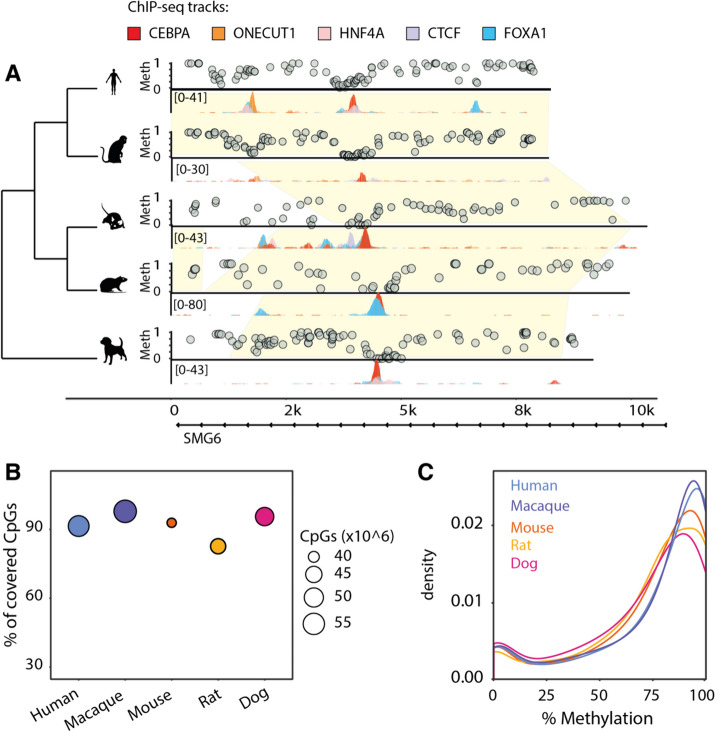
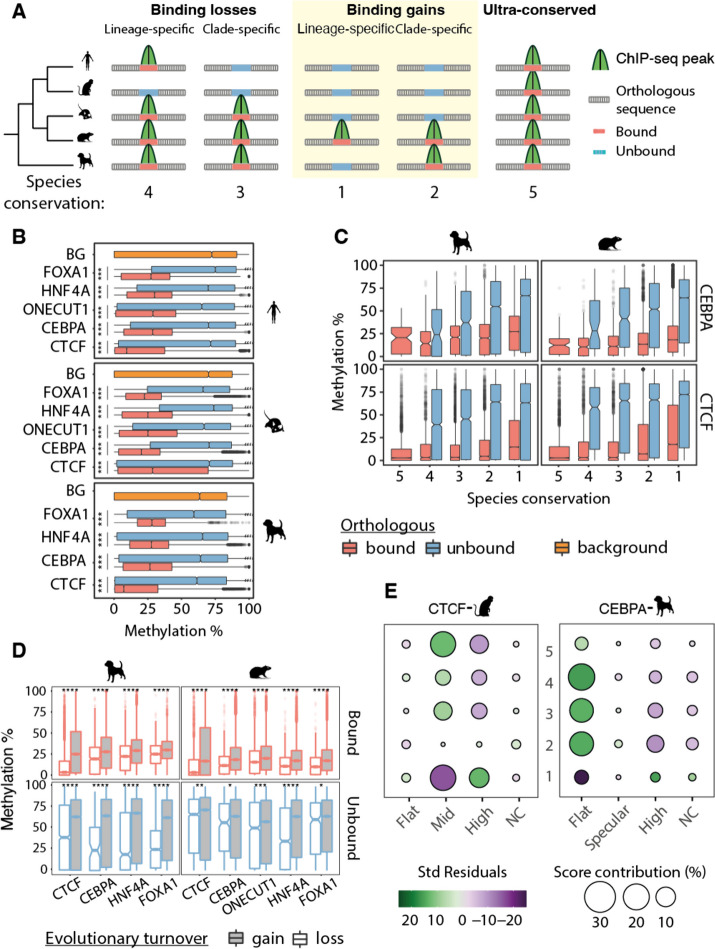
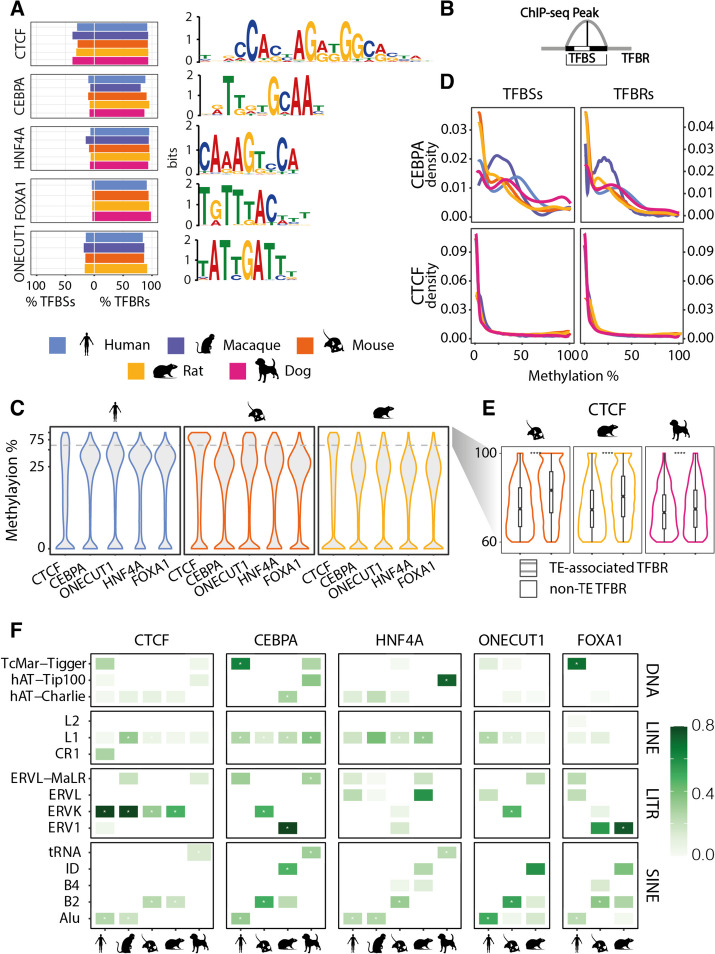
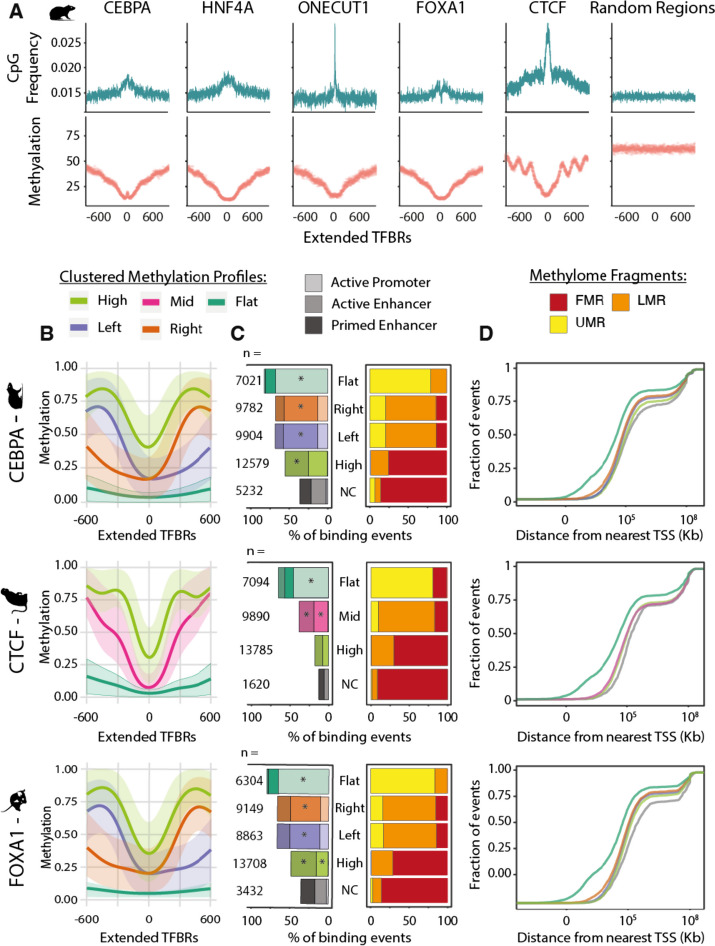
Here we use a comparative approach to investigate the association of DNA methylation to TF binding evolution in mammals. Specifically, we experimentally profile DNA methylation and combine this with published occupancy profiles of five distinct TFs (CTCF, CEBPA, HNF4A, ONECUT1, FOXA1) in the liver of five mammalian species (human, macaque, mouse, rat, dog). TF binding sites are lowly methylated, but they often also have intermediate methylation levels. Furthermore, biding sites are influenced by the methylation status of CpGs in their wider binding regions even when CpGs are absent from the core binding motif. Employing a classification and clustering approach, we extract distinct and species-conserved patterns of DNA methylation levels at TF binding regions. CEBPA, HNF4A, ONECUT1, and FOXA1 share the same methylation patterns, while CTCF's differ. These patterns characterize alternative functions and chromatin landscapes of TF-bound regions. Leveraging our phylogenetic framework, we find DNA methylation gain upon evolutionary loss of TF occupancy, indicating coordinated evolution. Furthermore, each methylation pattern has its own evolutionary trajectory reflecting its genomic contexts.
Our epigenomic analyses indicate a role for DNA methylation in TF binding changes across species including that specific DNA methylation profiles characterize TF binding and are associated with their regulatory activity, chromatin contexts, and evolutionary trajectories.
DNA 甲基化是一种重要的表观遗传修饰,在调节基因组功能方面具有多种作用。其水平在整个基因组中具有空间相关性,通常在抑制区域中较高,但在转录因子(TF)结合位点和活性调节区域中较低。然而,建立全基因组和 TF 结合位点甲基化模式的机制尚不清楚。
在这里,我们使用比较方法来研究哺乳动物中 DNA 甲基化与 TF 结合进化的相关性。具体来说,我们实验性地分析了 DNA 甲基化,并将其与五种类人动物(人类、猕猴、小鼠、大鼠、狗)肝脏中五个不同 TF(CTCF、CEBPA、HNF4A、ONECUT1、FOXA1)的已发表占据谱相结合。TF 结合位点的甲基化程度较低,但它们通常也具有中等甲基化水平。此外,即使核心结合基序中不存在 CpG,结合位点也会受到其更广泛的结合区域中 CpG 甲基化状态的影响。通过采用分类和聚类方法,我们提取了 TF 结合区域中 DNA 甲基化水平的独特且物种保守的模式。CEBPA、HNF4A、ONECUT1 和 FOXA1 具有相同的甲基化模式,而 CTCF 则不同。这些模式特征描述了 TF 结合区域的替代功能和染色质景观。利用我们的系统发育框架,我们发现 TF 占据丧失后 DNA 甲基化获得,表明协调进化。此外,每种甲基化模式都有自己的进化轨迹,反映了其基因组背景。
我们的表观基因组分析表明,DNA 甲基化在 TF 结合在不同物种中的变化中起作用,包括特定的 DNA 甲基化模式特征描述 TF 结合,并与它们的调节活性、染色质背景和进化轨迹相关。