Plant Germplasm and Genomics Center, Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, 650201, China.
College of Life Science and Agronomy, Zhoukou Normal University, Zhoukou, 466001, China.
Sci Rep. 2017 Sep 14;7(1):11546. doi: 10.1038/s41598-017-11367-x.
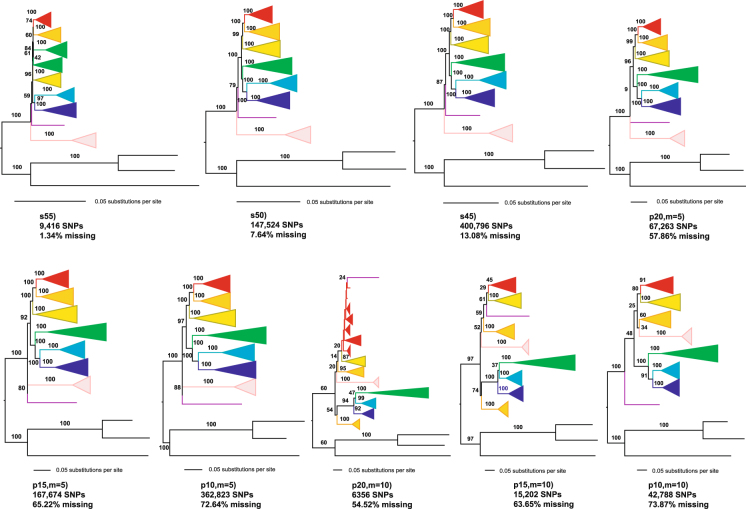
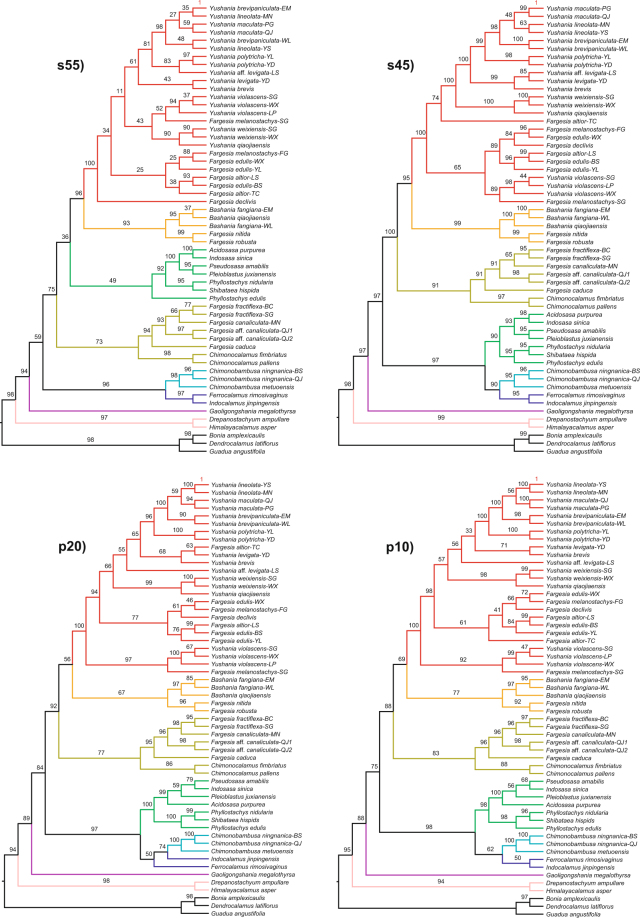
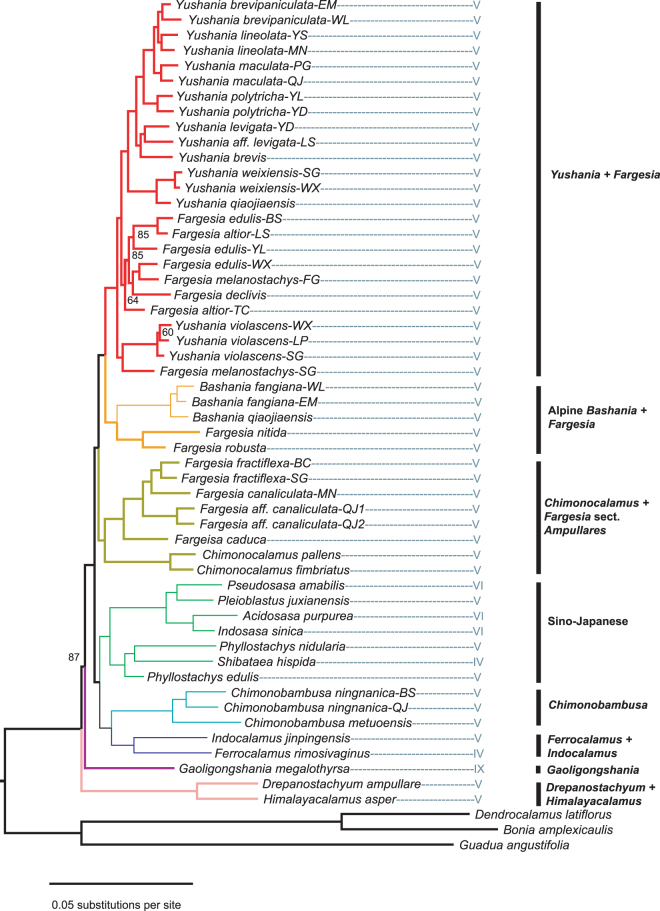
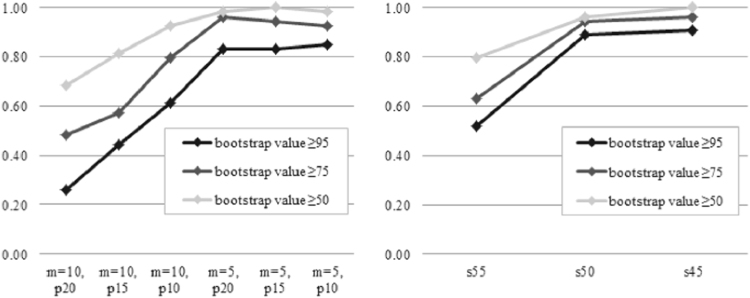
The temperate bamboos (tribe Arundinarieae, Poaceae) are strongly supported as monophyly in recent molecular studies, but taxonomic delineation and phylogenetic relationships within the tribe lack resolution. Here, we sampled 39 species (36 temperate bamboos and 3 outgroups) for restriction-site associated DNA sequencing (RAD-seq) with an emphasis on Phyllostachys clade and related clades. Using the largest data matrix for the bamboos to date, we were able to infer phylogenetic relationships with unparalleled resolution. The Phyllostachys, Shibataea, and Arundinaria clades defined from plastid phylogeny, were not supported as monophyletic group. However, the RAD-seq phylogeny largely agreed with the morphology-based taxonomy, with two clades having leptomorph rhizomes strongly supported as monophyletic group. We also explored two approaches, BWA-GATK (a mapping system) and Stacks (a grouping system), for differences in SNP calling and phylogeny inference. For the same level of missing data, the BWA-GATK pipeline produced much more SNPs in comparison with Stacks. Phylogenetic analyses of the largest data matrices from both pipelines, using concatenation and coalescent methods provided similar tree topologies, despite the presence of missing data. Our study demonstrates the utility of RAD-seq data for elucidating phylogenetic relationships between genera and higher taxonomic levels in this important but phylogenetically challenging group.
温带竹类(竹亚科,禾本科)在最近的分子研究中被强烈支持为单系群,但该亚科内的分类划界和系统发育关系仍缺乏分辨率。在这里,我们对 39 种物种(36 种温带竹子和 3 个外类群)进行了基于限制位点相关 DNA 测序(RAD-seq)的采样,重点是刚竹属和相关类群。利用迄今为止最大的竹子数据集,我们能够以无与伦比的分辨率推断出系统发育关系。基于叶绿体基因组构建的刚竹属、矢竹属和大明竹属分支并未被支持为单系群。然而,RAD-seq 系统发育树在很大程度上与基于形态的分类学一致,有两个具瘦形根茎的分支被强烈支持为单系群。我们还探索了两种方法,BWA-GATK(一种映射系统)和 Stacks(一种分组系统),用于 SNP 调用和系统发育推断的差异。在相同缺失数据水平下,BWA-GATK 与 Stacks 相比,生成了更多的 SNP。尽管存在缺失数据,但使用两种方法(最大数据集的连锁和合并分析方法)对来自两个管道的最大数据集进行的系统发育分析提供了相似的树拓扑结构。我们的研究表明,RAD-seq 数据可用于阐明该重要但系统发育上具有挑战性的类群中属间和更高分类群之间的系统发育关系。