Yang Jinliang, Mezmouk Sofiane, Baumgarten Andy, Buckler Edward S, Guill Katherine E, McMullen Michael D, Mumm Rita H, Ross-Ibarra Jeffrey
Department of Plant Sciences, University of California, Davis, Davis, California, United States of America.
DuPont Pioneer, Johnston, Iowa, United States of America.
PLoS Genet. 2017 Sep 27;13(9):e1007019. doi: 10.1371/journal.pgen.1007019. eCollection 2017 Sep.
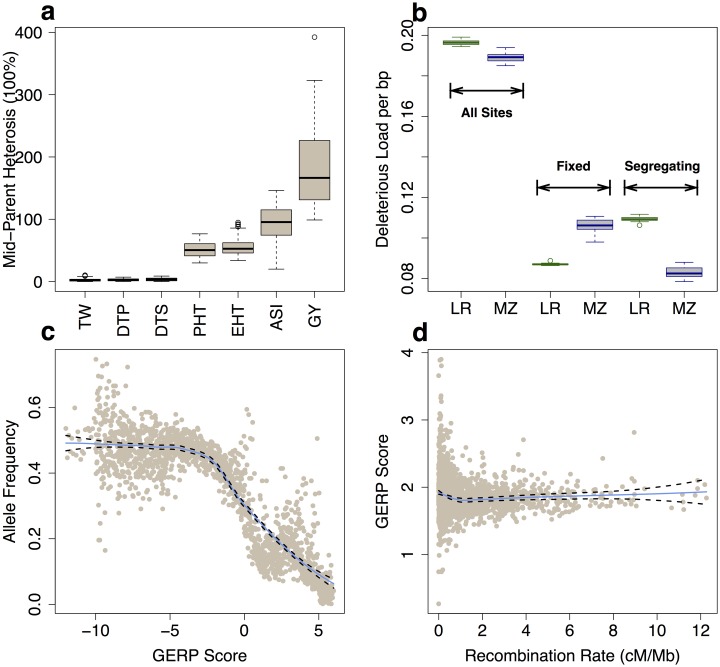
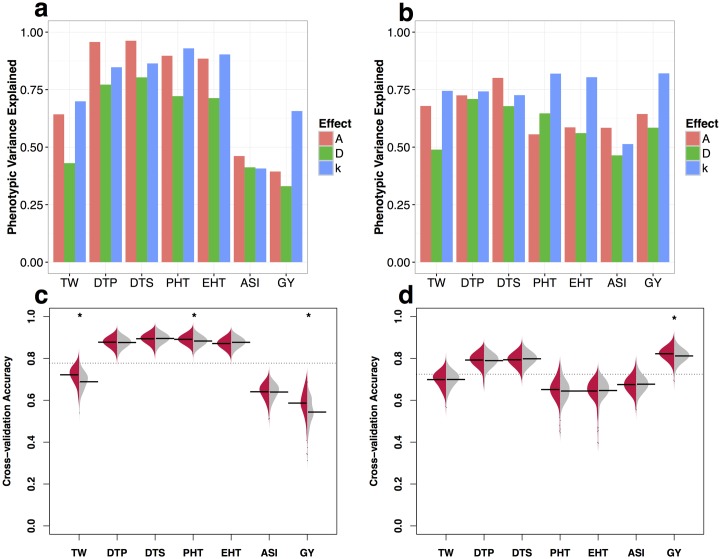
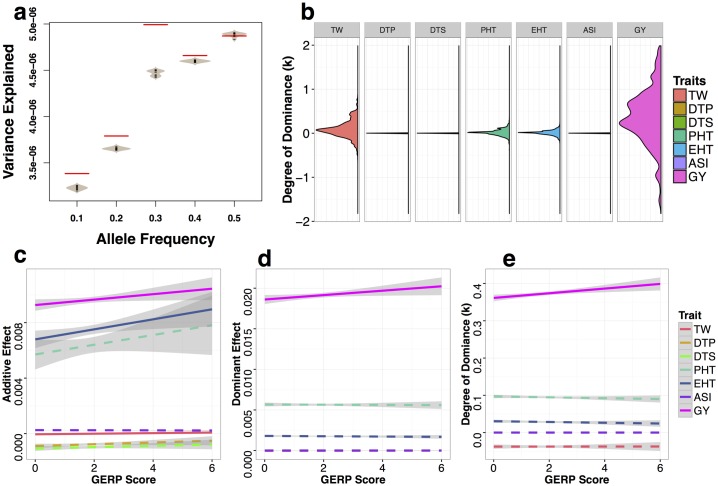
Deleterious alleles have long been proposed to play an important role in patterning phenotypic variation and are central to commonly held ideas explaining the hybrid vigor observed in the offspring of a cross between two inbred parents. We test these ideas using evolutionary measures of sequence conservation to ask whether incorporating information about putatively deleterious alleles can inform genomic selection (GS) models and improve phenotypic prediction. We measured a number of agronomic traits in both the inbred parents and hybrids of an elite maize partial diallel population and re-sequenced the parents of the population. Inbred elite maize lines vary for more than 350,000 putatively deleterious sites, but show a lower burden of such sites than a comparable set of traditional landraces. Our modeling reveals widespread evidence for incomplete dominance at these loci, and supports theoretical models that more damaging variants are usually more recessive. We identify haplotype blocks using an identity-by-decent (IBD) analysis and perform genomic prediction analyses in which we weigh blocks on the basis of complementation for segregating putatively deleterious variants. Cross-validation results show that incorporating sequence conservation in genomic selection improves prediction accuracy for grain yield and other fitness-related traits as well as heterosis for those traits. Our results provide empirical support for an important role for incomplete dominance of deleterious alleles in explaining heterosis and demonstrate the utility of incorporating functional annotation in phenotypic prediction and plant breeding.
长期以来,人们一直认为有害等位基因在塑造表型变异方面发挥着重要作用,并且是解释两个自交亲本杂交后代中观察到的杂种优势这一普遍观点的核心。我们使用序列保守性的进化指标来检验这些观点,以探讨纳入关于假定有害等位基因的信息是否能为基因组选择(GS)模型提供信息并改善表型预测。我们测量了一个优良玉米部分双列杂交群体的自交亲本和杂种的多个农艺性状,并对该群体的亲本进行了重测序。自交优良玉米品系在超过350,000个假定有害位点上存在差异,但与一组可比的传统地方品种相比,此类位点的负担较低。我们的建模揭示了这些位点存在广泛的不完全显性证据,并支持理论模型,即更具破坏性的变异通常更隐性。我们使用基于血缘一致性(IBD)分析来识别单倍型块,并进行基因组预测分析,在分析中我们根据分离假定有害变异的互补性对块进行加权。交叉验证结果表明,在基因组选择中纳入序列保守性可提高对籽粒产量和其他适应性相关性状的预测准确性,以及这些性状的杂种优势。我们的结果为有害等位基因的不完全显性在解释杂种优势中的重要作用提供了实证支持,并证明了在表型预测和植物育种中纳入功能注释的实用性。