Department of Chemistry, Yale University , New Haven, Connecticut 06520, United States.
Department of Pharmacology, Yale School of Medicine , New Haven, Connecticut 06520, United States.
Acc Chem Res. 2017 Oct 17;50(10):2577-2588. doi: 10.1021/acs.accounts.7b00347. Epub 2017 Sep 28.
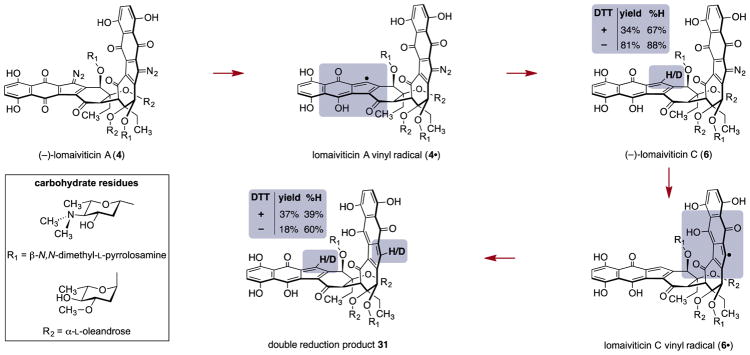
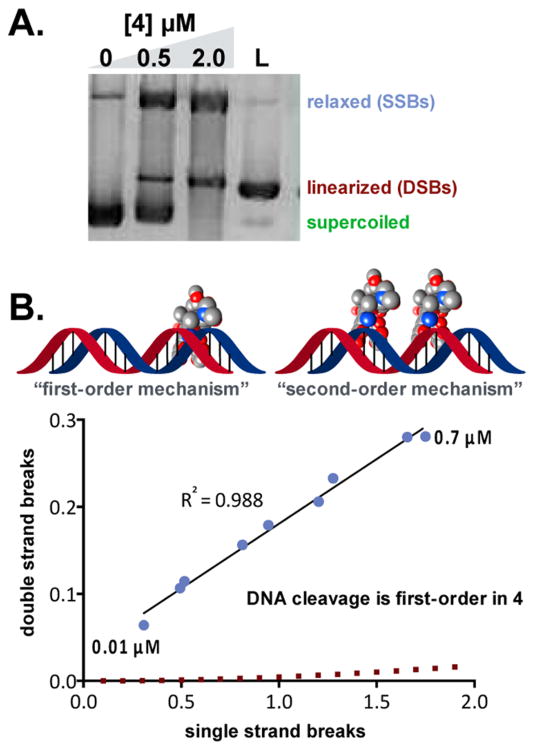
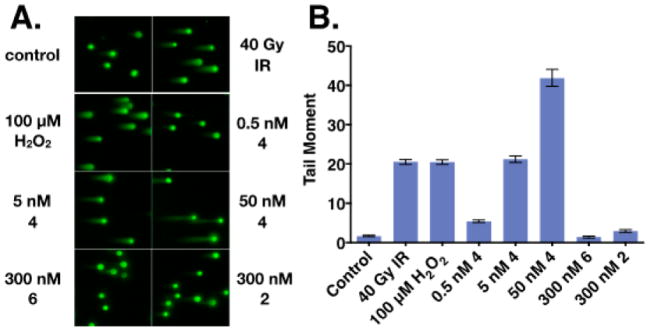
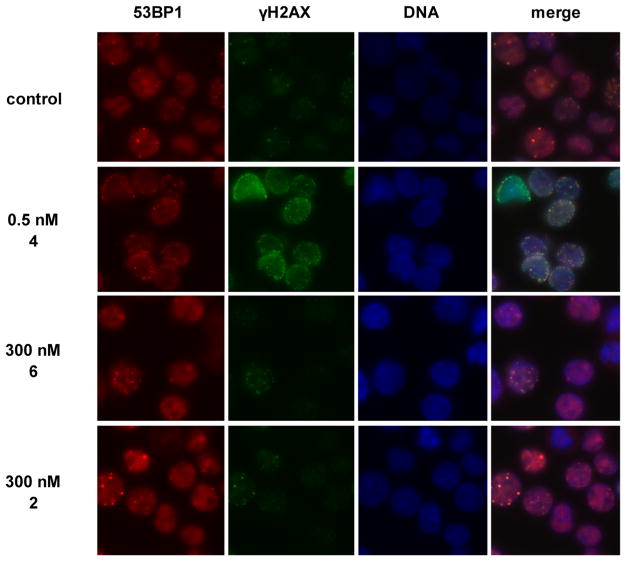
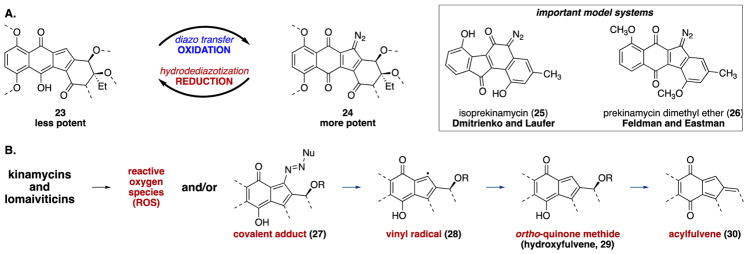
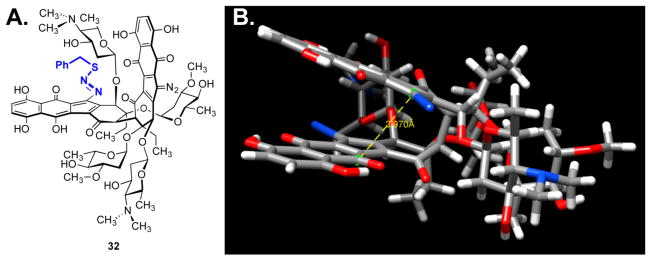
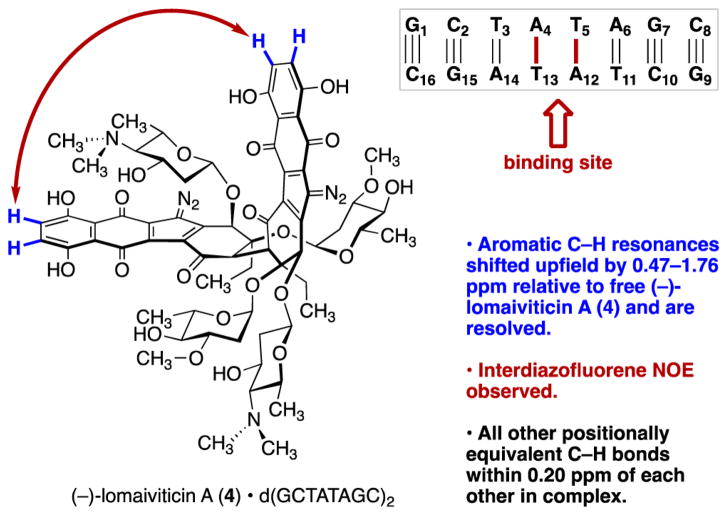
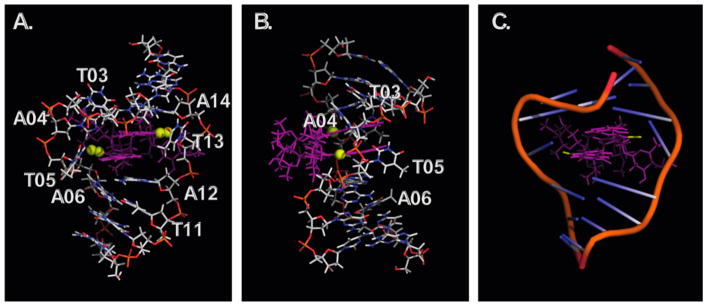
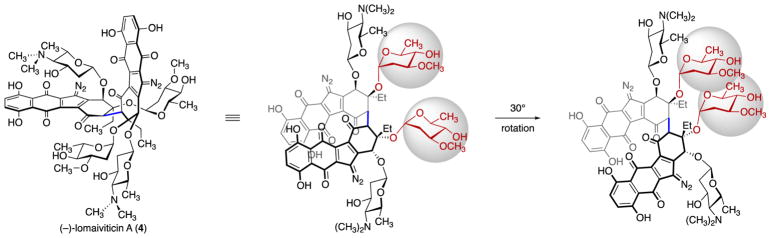
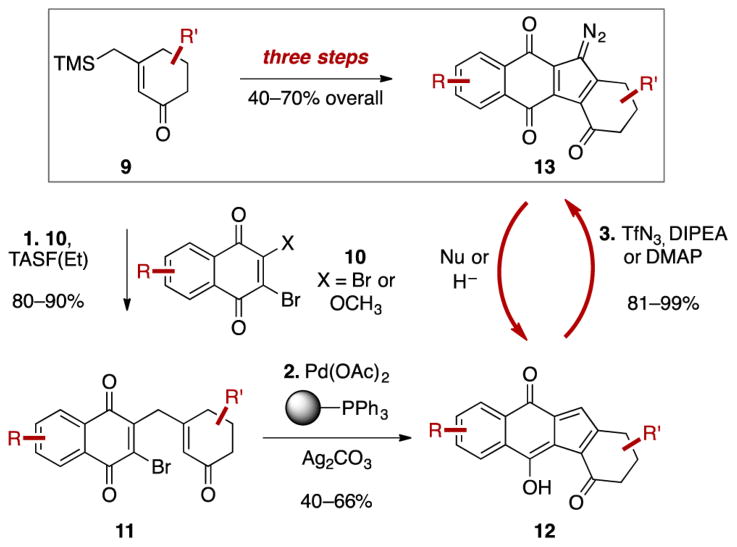
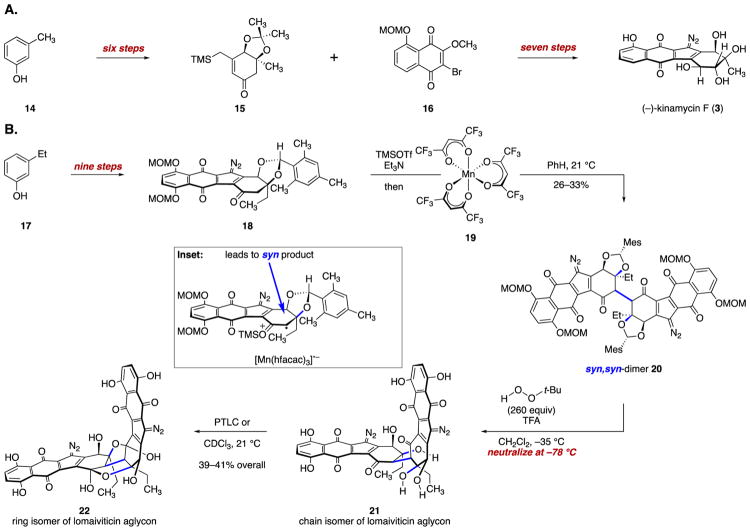
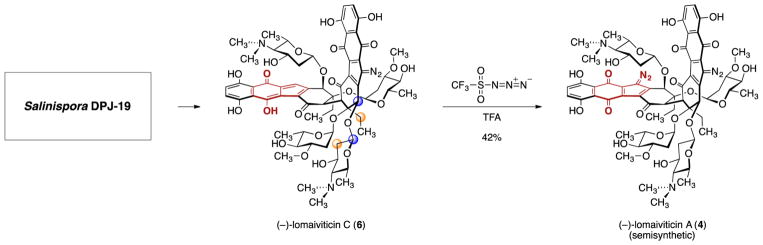
(-)-Lomaiviticin A (4) is a complex C-symmetric bacterial metabolite that contains two diazofluorene functional groups. The diazofluorene consists of naphthoquinone, cyclopentadiene, and diazo substituents fused through a σ- and π-bonding network. Additionally, (-)-lomaiviticin A (4) is a potent cytotoxin, with half-maximal inhibitory potency (IC) values in the low nanomolar range against many cancer cell lines. Because of limitations in supply, its mechanism of action had remained a "black box" since its isolation in the early 2000s. In this Account, I describe how studies directed toward the total synthesis of (-)-lomaiviticin A (4) provided a platform to elucidate the emergent properties of this metabolite and thereby connect chemical reactivity with cellular phenotype. We first developed a convergent strategy to prepare the diazofluorene (9 + 10 → 13). We then adapted this chemistry to the synthesis of lomaiviticin aglycon (21/22) and the natural monomeric diazofluorene (-)-kinamycin F (3). The key step in the lomaiviticin aglycon (21/22) synthesis involved the stereoselective oxidative coupling of two monomeric diazofluorenes (2 × 18 → 20) to establish the cojoining carbon-carbon bond of the target. As the absolute stereochemistry of the aglycon and carbohydrate residues of (-)-lomaiviticin A (4) were unknown, we developed a semisynthetic route to the metabolite that proceeds in one step and 42% yield by diazo transfer to the more abundant isolate (-)-lomaiviticin C (6). This allowed us to complete the stereochemical assignment of (-)-lomaiviticin A (4) and provided a renewable source of material. Using this material, we established that the remarkable cytotoxic effects of (-)-lomaiviticin A (4) derive from the induction of highly toxic double-strand breaks (DSBs) in DNA. At the molecular level, 1,7-nucleophilic additions to each electrophilic diazofluorene trigger homolytic decomposition pathways that produce sp radicals at the carbon atoms of each diazo group. These radicals abstract hydrogen atoms from the deoxyribose of DNA, a process known to initiate strand cleavage. NMR spectroscopy and molecular mechanics simulations were used to elucidate the mode of DNA binding. These studies showed that both diazofluorenes of (-)-lomaiviticin A (4) penetrate into the duplex. This mode of non-covalent binding places each diazo carbon atom in close proximity to each DNA strand. Throughout these studies, isolates containing one diazofluorene, such as (-)-lomaiviticin C (6) and (-)-kinamycin C (2), were used as controls. Consistent with our mechanistic model, these compounds do not induce DSBs in DNA and are several orders of magnitude less potent. Reactivity studies suggest that (-)-lomaiviticin A (4) is more electrophilic than simple monomeric diazofluorenes. We attribute this to through-space delocalization of the developing negative charge in the transition state for 1,7-addition. Consistent with this mechanism of action, (-)-lomaiviticin A (4) displays selective low-picomolar potencies toward DNA DSB repair-deficient cell types. The emergent properties of (-)-lomaiviticin A (4) derive from the specific arrangement of diazo, naphthoquinone, cyclopentadiene, and ketone functional groups. These functional groups work together to yield, essentially, a masked vinyl radical that can be exposed under biological conditions. Furthermore, the rotational symmetry of the metabolite, deriving from dimerization, allows it to interact with the antiparallel symmetry of DNA and affect cleavage of the duplex.
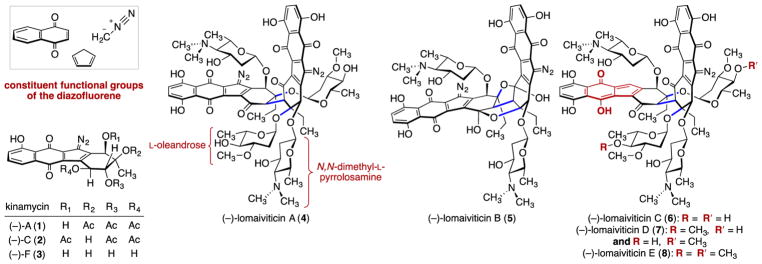
(-)-洛马维定 A(4)是一种复杂的 C 轴对称细菌代谢产物,含有两个重氮氟苯官能团。该重氮氟苯由萘醌、环戊二烯和重氮取代基通过 σ 和 π 键合网络融合而成。此外,(-)-洛马维定 A(4)是一种有效的细胞毒素,对许多癌细胞系的半数最大抑制效力(IC)值在纳摩尔范围内。由于供应有限,自 21 世纪初分离以来,其作用机制一直是一个“黑匣子”。在本报告中,我描述了如何针对(-)-洛马维定 A(4)的全合成进行研究,为阐明该代谢产物的新兴特性提供了一个平台,并由此将化学反应性与细胞表型联系起来。我们首先开发了一种会聚策略来制备重氮氟苯(9+10→13)。然后,我们将这种化学方法应用于洛马维定苷(21/22)和天然单体重氮氟苯(-)kinamycin F(3)的合成。洛马维定苷(21/22)合成中的关键步骤涉及两个单体重氮氟苯(2×18→20)的立体选择性氧化偶联,以建立目标的共连接碳-碳键。由于(-)-洛马维定 A(4)的糖苷和碳水化合物残基的绝对立体化学结构未知,我们开发了一种半合成途径,可以一步以 42%的产率通过重氮转移到更丰富的分离物(-)洛马维定 C(6)来完成代谢物的合成。这使我们能够完成(-)-洛马维定 A(4)的立体化学分配,并提供了可再生的材料来源。使用这种材料,我们确定了(-)-洛马维定 A(4)的显著细胞毒性作用源自 DNA 中高度有毒的双链断裂(DSB)的诱导。在分子水平上,每个亲电重氮氟苯的 1,7-亲核加成引发均裂分解途径,在每个重氮基团的碳原子上产生 sp 自由基。这些自由基从 DNA 的脱氧核糖中提取氢原子,这一过程被认为是引发链断裂的过程。NMR 光谱和分子力学模拟被用来阐明 DNA 结合的模式。这些研究表明,(-)-洛马维定 A(4)的两个重氮氟苯都穿透到双链中。这种非共价结合模式使每个重氮碳原子都与 DNA 链紧密接近。在这些研究中,包含一个重氮氟苯的分离物,如(-)洛马维定 C(6)和(-)kinamycin C(2),被用作对照。与我们的机制模型一致,这些化合物不会在 DNA 中诱导 DSBs,并且效力低几个数量级。反应性研究表明,(-)-洛马维定 A(4)比简单的单体重氮氟苯更具亲电性。我们将其归因于 1,7-加成过渡态中发展的负电荷的空间离域。与这种作用机制一致,(-)-洛马维定 A(4)对 DNA DSB 修复缺陷细胞类型显示出选择性的低皮摩尔效力。(-)-洛马维定 A(4)的新兴特性源于重氮、萘醌、环戊二烯和酮官能团的特定排列。这些官能团协同作用,基本上产生了一个可以在生物条件下暴露的掩蔽乙烯基自由基。此外,代谢物的旋转对称性,源于二聚化,使其能够与 DNA 的反平行对称性相互作用并影响双链的切割。