UCL Huntington's Disease Centre, Sobell Department of Motor Neuroscience, UCL Institute of Neurology, University College London, London, United Kingdom.
Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, 02139, United States.
Sci Rep. 2017 Oct 2;7(1):12556. doi: 10.1038/s41598-017-12897-0.
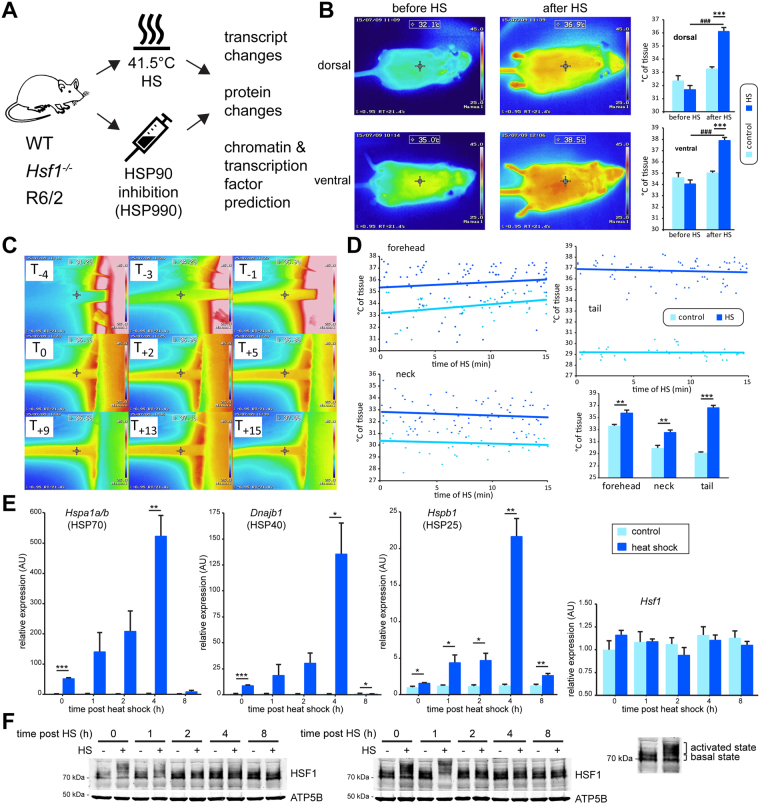
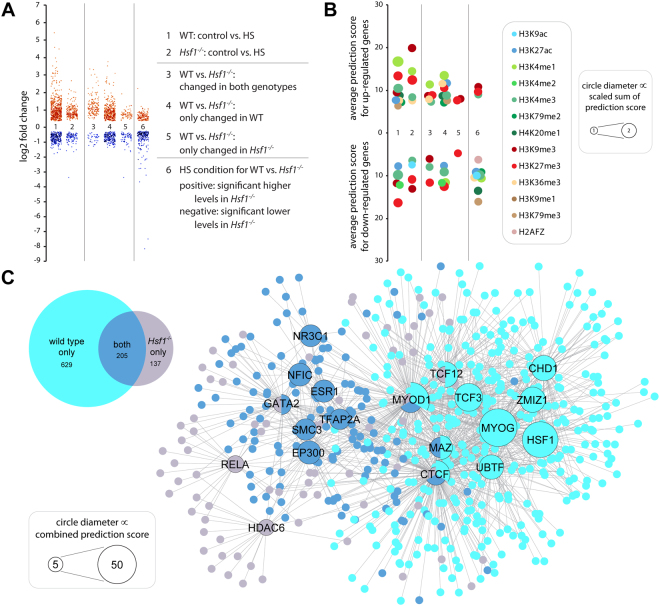
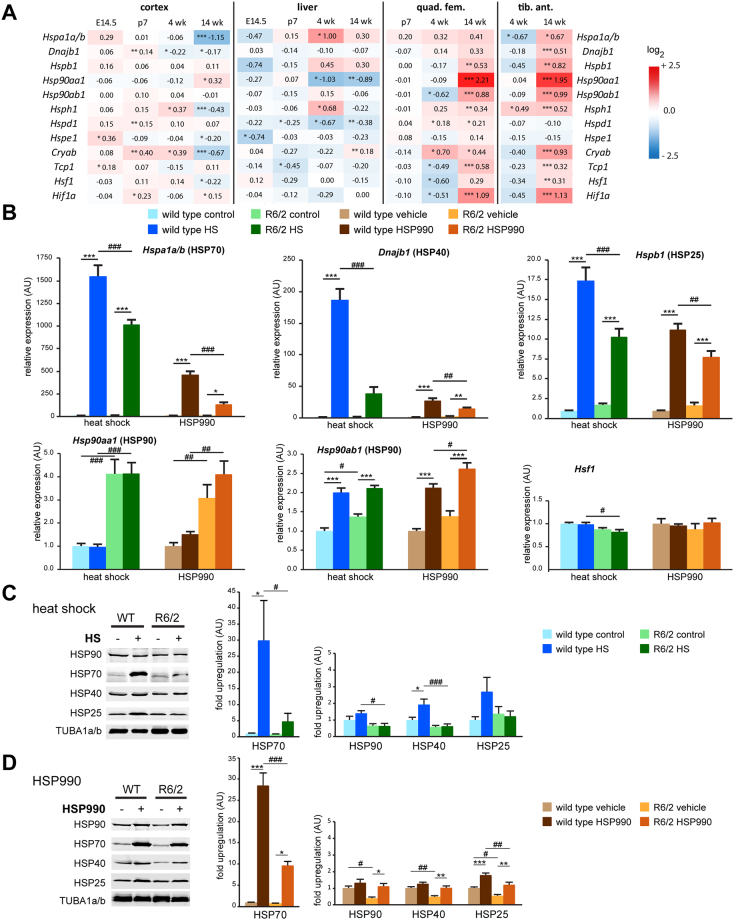
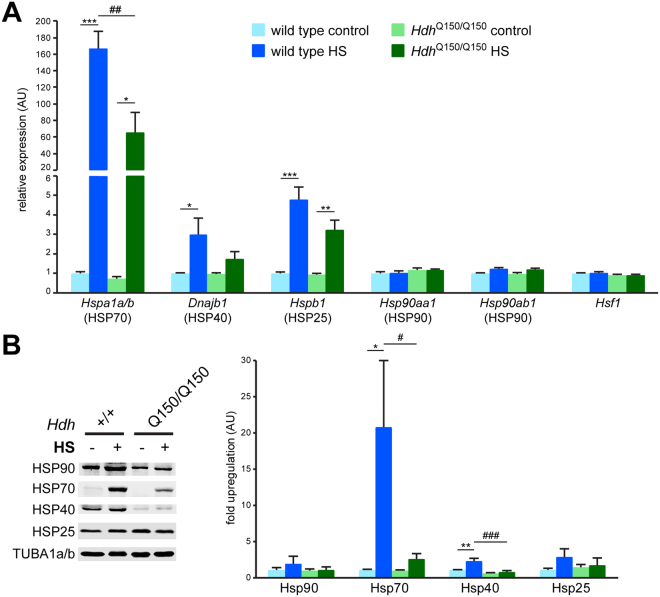
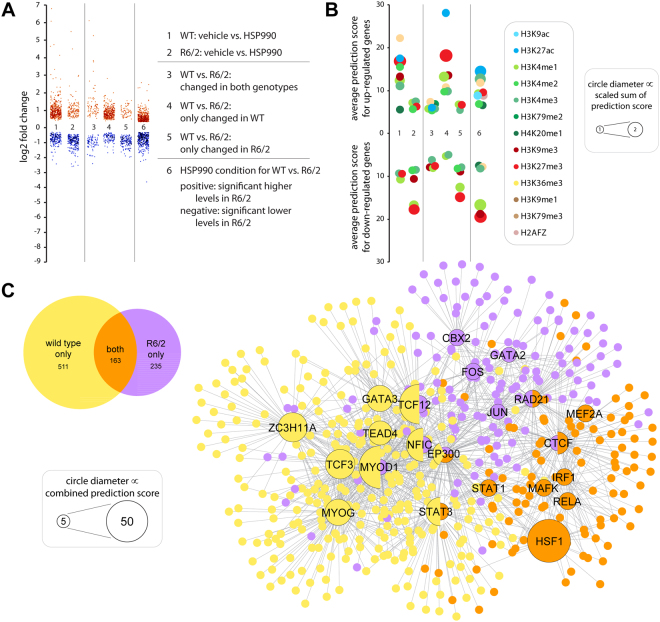
The heat shock response (HSR) is a mechanism to cope with proteotoxic stress by inducing the expression of molecular chaperones and other heat shock response genes. The HSR is evolutionarily well conserved and has been widely studied in bacteria, cell lines and lower eukaryotic model organisms. However, mechanistic insights into the HSR in higher eukaryotes, in particular in mammals, are limited. We have developed an in vivo heat shock protocol to analyze the HSR in mice and dissected heat shock factor 1 (HSF1)-dependent and -independent pathways. Whilst the induction of proteostasis-related genes was dependent on HSF1, the regulation of circadian function related genes, indicating that the circadian clock oscillators have been reset, was independent of its presence. Furthermore, we demonstrate that the in vivo HSR is impaired in mouse models of Huntington's disease but we were unable to corroborate the general repression of transcription that follows a heat shock in lower eukaryotes.
热休克反应 (HSR) 是一种通过诱导分子伴侣和其他热休克反应基因的表达来应对蛋白毒性应激的机制。HSR 在进化上高度保守,并在细菌、细胞系和较低等真核模式生物中得到了广泛研究。然而,对于高等真核生物(特别是哺乳动物)中 HSR 的机制性认识仍然有限。我们开发了一种体内热休克方案来分析小鼠中的 HSR,并剖析了热休克因子 1 (HSF1) 依赖和非依赖途径。虽然与蛋白质稳态相关的基因的诱导依赖于 HSF1,但与生物钟功能相关的基因的调控,表明生物钟振荡器已经被重置,却不依赖于 HSF1 的存在。此外,我们证明亨廷顿病的小鼠模型中的体内 HSR 受损,但我们无法证实在较低等真核生物中热休克后转录的普遍抑制。