Laurita Geneva, Fabini Douglas H, Stoumpos Constantinos C, Kanatzidis Mercouri G, Seshadri Ram
Materials Research Laboratory , University of California , Santa Barbara , California 93106 , USA.
Materials Department , University of California , Santa Barbara , California 93106 , USA.
Chem Sci. 2017 Aug 1;8(8):5628-5635. doi: 10.1039/c7sc01429e. Epub 2017 Jun 16.
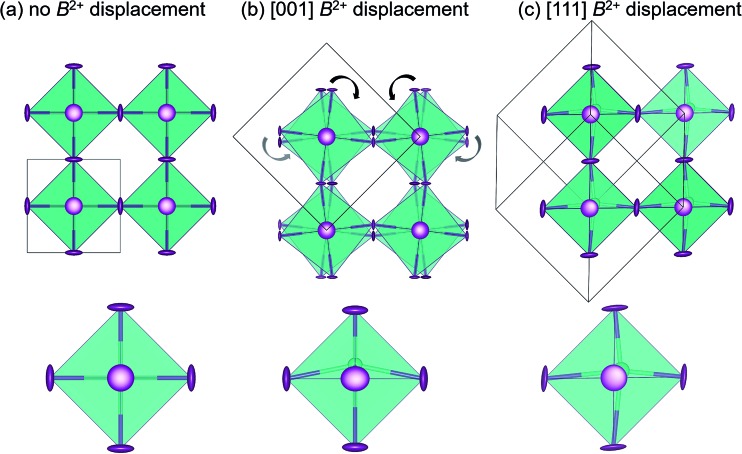
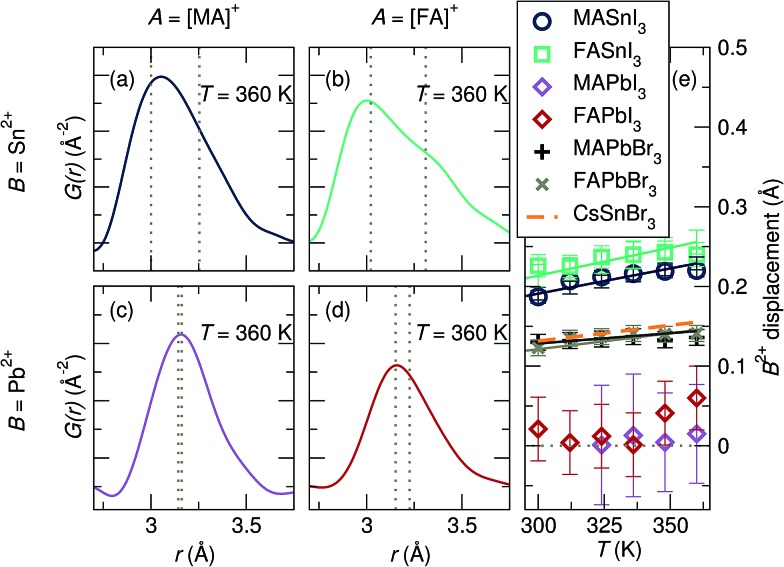
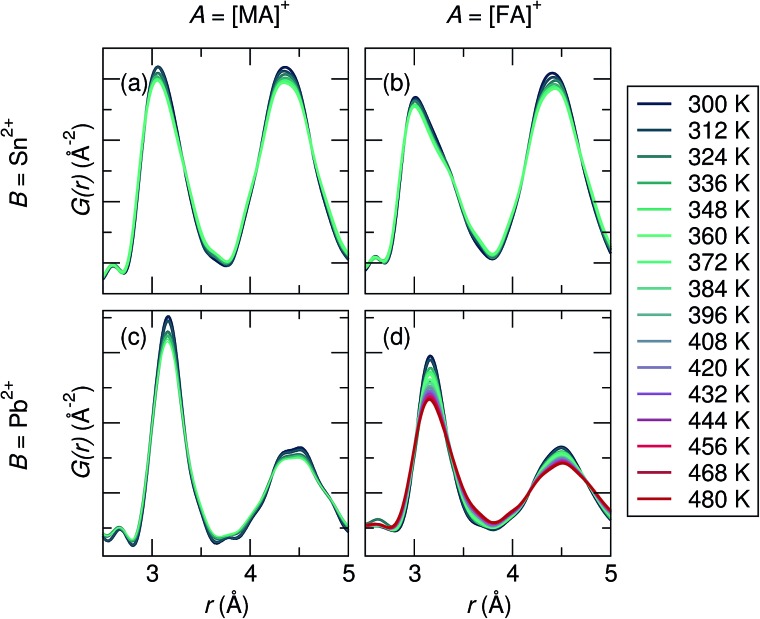
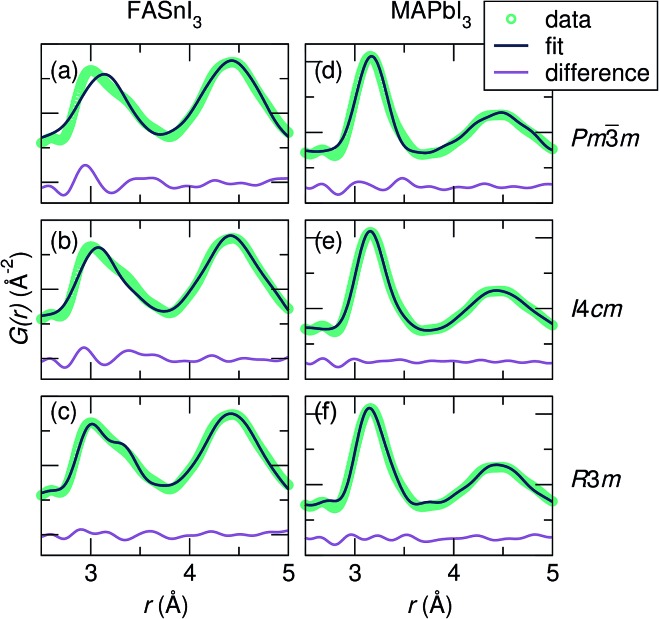
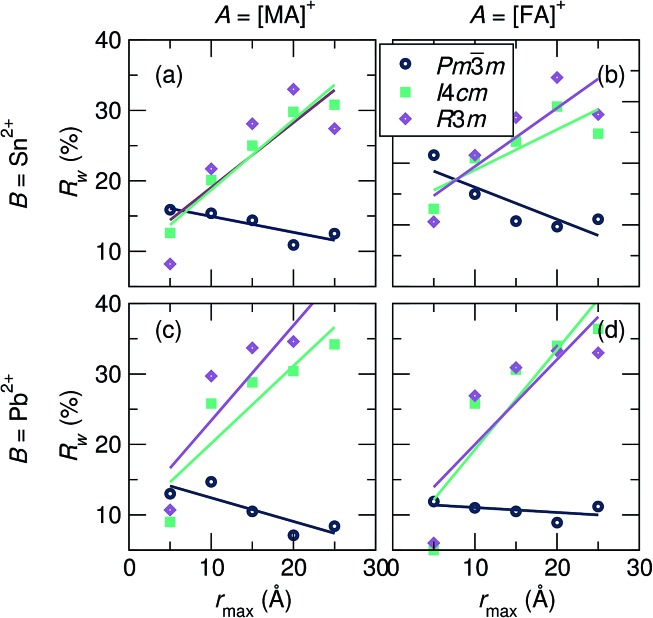
Hybrid halide perovskites combine ease of preparation and relatively abundant constituent elements with fascinating photophysical properties. Descriptions of the chemical and structural drivers of the remarkable properties have often focused on the potential role of the dynamic order/disorder of the molecular A-site cations. We reveal here a key aspect of the inorganic framework that potentially impacts the electronic, thermal, and dielectric properties. The temperature evolution of the X-ray pair distribution functions of hybrid perovskites ABX [A = CHNH (MA) or CH(NH) (FA); B = Sn or Pb; X = Br, or I] in their cubic phases above 300 K reveals temperature-activated displacement (off-centering) of the divalent group 14 cations from their nominal, centered sites. This symmetry-lowering distortion phenomenon, previously dubbed in the context of compounds such as PbTe, is attributed to Sn and Pb lone pair stereochemistry. Of the materials studied here, the largest displacements from the center of the octahedral sites are found in tin iodides, a more moderate effect is found in lead bromides, and the weakest effect is seen in lead iodides. The A-site cation appears to play a role as well, with the larger FA resulting in greater off-centering for both Sn and Pb. Dynamic off-centering, which is concealed within the framework of traditional Bragg crystallography, is proposed to play a key role in the remarkable defect-tolerant nature of transport in these semiconductors its effect on the polarizability of the lattice. The results suggest a novel chemical design principle for future materials discovery.
混合卤化物钙钛矿兼具易于制备、组成元素相对丰富以及迷人的光物理性质。对这些卓越性质的化学和结构驱动因素的描述,常常聚焦于分子A位阳离子动态有序/无序的潜在作用。我们在此揭示了无机骨架的一个关键方面,它可能会影响电子、热和介电性质。混合钙钛矿ABX[A = CH₃NH₃⁺(MA)或CH(NH₂)₂⁺(FA); B = Sn或Pb; X = Br或I]在300 K以上立方相中的X射线对分布函数的温度演化,揭示了二价14族阳离子从其标称的中心位置发生温度激活位移(偏离中心)。这种降低对称性的畸变现象,在诸如PbTe等化合物的背景下之前就有提及,归因于Sn和Pb的孤对立体化学。在此研究的材料中,八面体位置中心最大的位移出现在碘化锡中,在溴化铅中发现的效应较为适中,而在碘化铅中效应最弱。A位阳离子似乎也起到了作用,较大的FA导致Sn和Pb的偏离中心程度更大。动态偏离中心隐藏在传统布拉格晶体学的框架内,被认为在这些半导体显著的缺陷容忍输运性质中起关键作用——其对晶格极化率的影响。这些结果为未来材料发现提出了一种新颖的化学设计原则。