Fosso Bruno, Pesole Graziano, Rosselló Francesc, Valiente Gabriel
1 Institute of Biomembranes and Bioenergetics , Consiglio Nazionale delle Ricerche, Bari, Italy .
2 Department of Mathematics and Computer Science, Balearic Islands Health Research Institute (IdISBa), University of the Balearic Islands , Palma de Mallorca, Spain .
J Comput Biol. 2018 Mar;25(3):348-360. doi: 10.1089/cmb.2017.0144. Epub 2017 Oct 13.
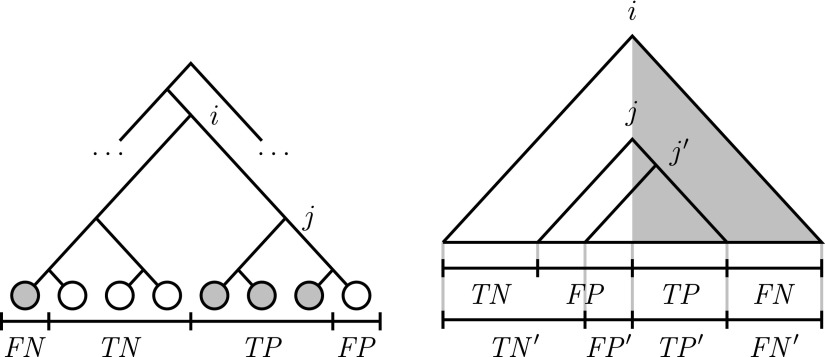
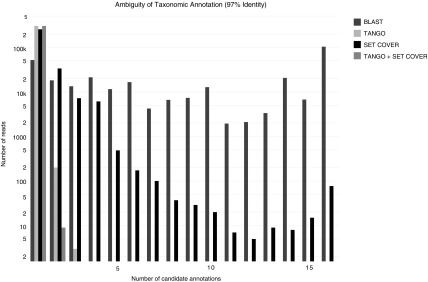
The classification of reads from a metagenomic sample using a reference taxonomy is usually based on first mapping the reads to the reference sequences and then classifying each read at a node under the lowest common ancestor of the candidate sequences in the reference taxonomy with the least classification error. However, this taxonomic annotation can be biased by an imbalanced taxonomy and also by the presence of multiple nodes in the taxonomy with the least classification error for a given read. In this article, we show that the Rand index is a better indicator of classification error than the often used area under the receiver operating characteristic (ROC) curve and F-measure for both balanced and imbalanced reference taxonomies, and we also address the second source of bias by reducing the taxonomic annotation problem for a whole metagenomic sample to a set cover problem, for which a logarithmic approximation can be obtained in linear time and an exact solution can be obtained by integer linear programming. Experimental results with a proof-of-concept implementation of the set cover approach to taxonomic annotation in a next release of the TANGO software show that the set cover approach further reduces ambiguity in the taxonomic annotation obtained with TANGO without distorting the relative abundance profile of the metagenomic sample.
使用参考分类法对宏基因组样本中的 reads 进行分类,通常是先将 reads 映射到参考序列,然后在参考分类法中候选序列的最低共同祖先下的节点处,以分类错误最小的方式对每个 reads 进行分类。然而,这种分类注释可能会受到不平衡分类法的影响,也会受到分类法中存在多个节点的影响,对于给定的 reads,这些节点的分类错误最小。在本文中,我们表明,对于平衡和不平衡的参考分类法,兰德指数比常用的受试者工作特征(ROC)曲线下面积和 F 度量更能作为分类错误的指标,并且我们还通过将整个宏基因组样本的分类注释问题简化为集合覆盖问题来解决第二个偏差来源,对于该问题,可以在线性时间内获得对数近似值,并通过整数线性规划获得精确解。在 TANGO 软件的下一个版本中,使用集合覆盖方法进行分类注释的概念验证实现的实验结果表明,集合覆盖方法在不扭曲宏基因组样本相对丰度分布的情况下,进一步减少了用 TANGO 获得的分类注释中的模糊性。