Swiss Federal Institute of Technology Zurich, Department of Biology, IMHS, Zurich, Switzerland.
Life Science Zurich Graduate School, Molecular Life Science program, University of Zürich, Switzerland.
Brief Bioinform. 2019 Jan 18;20(1):288-298. doi: 10.1093/bib/bbx115.
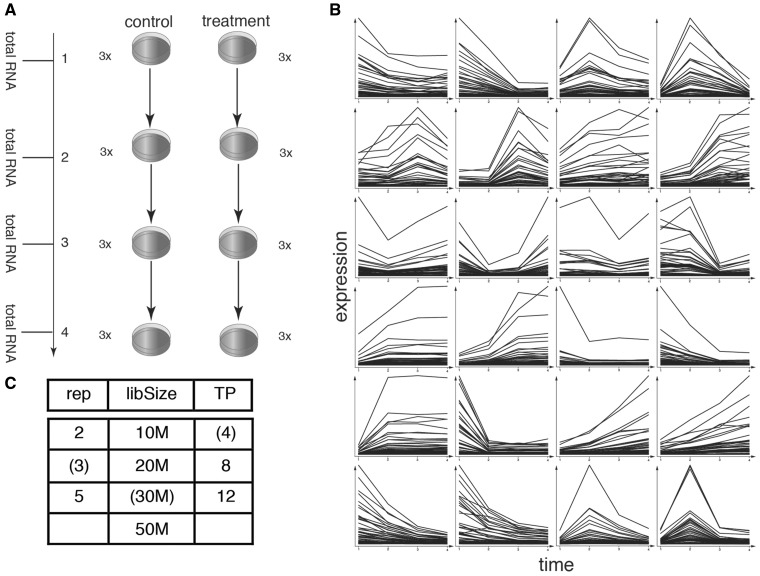
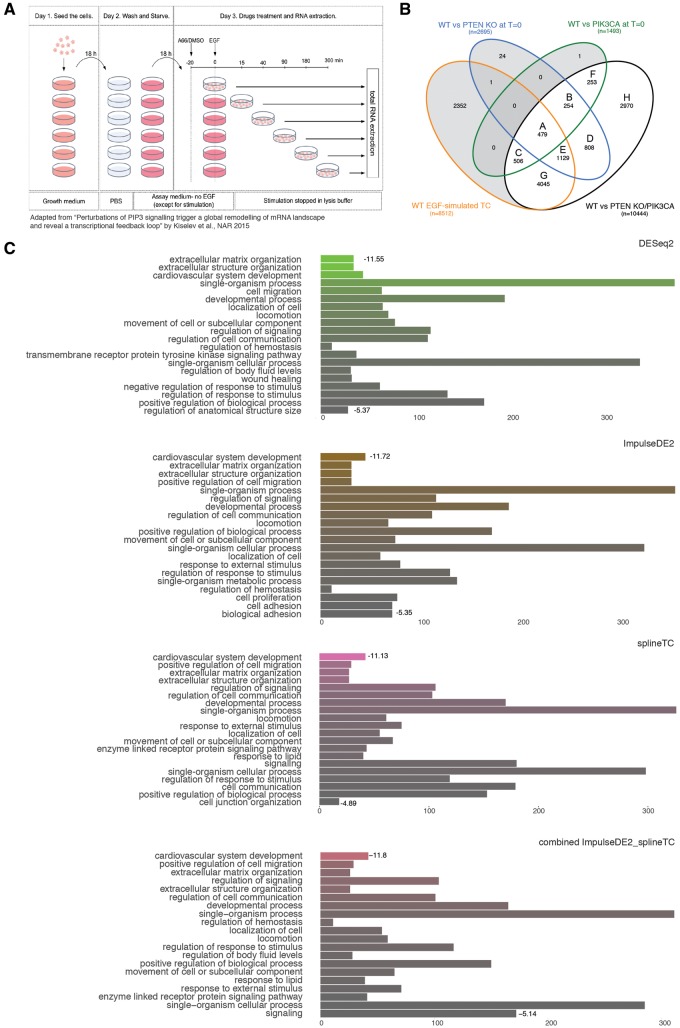
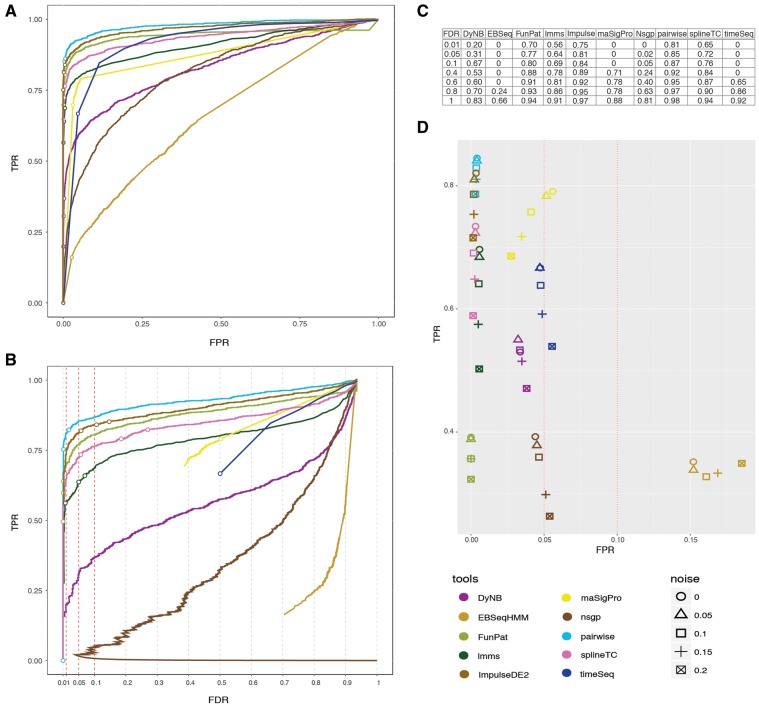
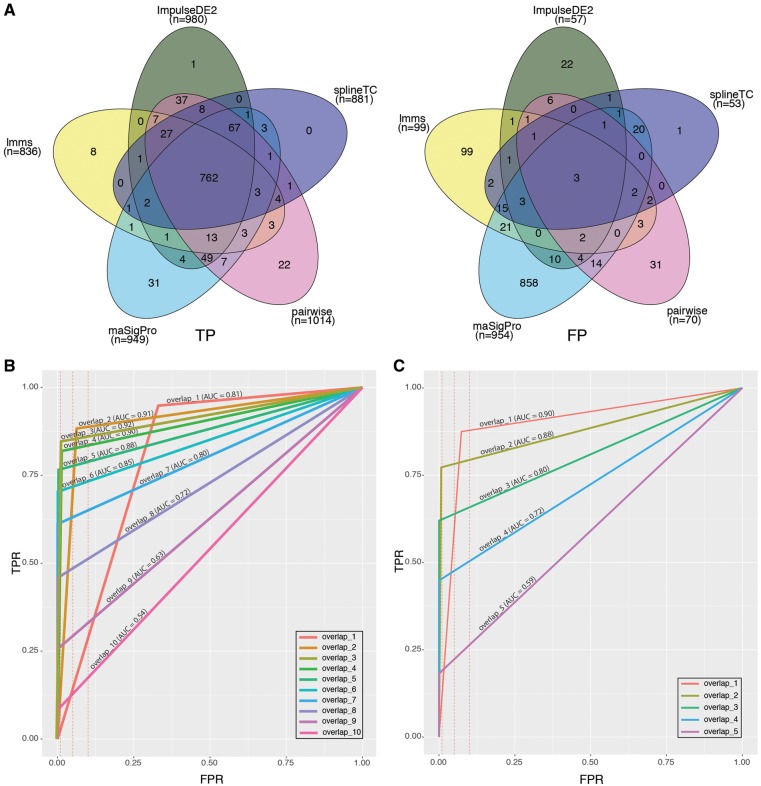
RNA sequencing (RNA-seq) has become a standard procedure to investigate transcriptional changes between conditions and is routinely used in research and clinics. While standard differential expression (DE) analysis between two conditions has been extensively studied, and improved over the past decades, RNA-seq time course (TC) DE analysis algorithms are still in their early stages. In this study, we compare, for the first time, existing TC RNA-seq tools on an extensive simulation data set and validated the best performing tools on published data. Surprisingly, TC tools were outperformed by the classical pairwise comparison approach on short time series (<8 time points) in terms of overall performance and robustness to noise, mostly because of high number of false positives, with the exception of ImpulseDE2. Overlapping of candidate lists between tools improved this shortcoming, as the majority of false-positive, but not true-positive, candidates were unique for each method. On longer time series, pairwise approach was less efficient on the overall performance compared with splineTC and maSigPro, which did not identify any false-positive candidate.
RNA 测序(RNA-seq)已成为研究条件间转录变化的标准程序,在研究和临床中经常使用。虽然已经对两种条件之间的标准差异表达(DE)分析进行了广泛研究,并在过去几十年中得到了改进,但 RNA-seq 时间序列(TC)DE 分析算法仍处于早期阶段。在这项研究中,我们首次在广泛的模拟数据集上比较了现有的 TC RNA-seq 工具,并在已发表的数据上验证了表现最好的工具。令人惊讶的是,TC 工具在短时间序列(<8 个时间点)方面的整体性能和对噪声的稳健性方面逊于经典的两两比较方法,这主要是由于假阳性数量过多,除了 ImpulseDE2 之外。工具之间的候选列表重叠改善了这一缺点,因为大多数假阳性候选者,但不是真正的阳性候选者,每个方法都是唯一的。在较长的时间序列上,与 splineTC 和 maSigPro 相比,两两比较方法在整体性能方面效率较低,后两者未识别出任何假阳性候选者。