Consiglio Arianna, Mencar Corrado, Grillo Giorgio, Marzano Flaviana, Caratozzolo Mariano Francesco, Liuni Sabino
Institute for Biomedical Technologies of Bari - ITB, National Research Council, Bari, 70126, Italy.
Department of Informatics, University of Bari Aldo Moro, Bari, 70121, Italy.
BMC Bioinformatics. 2016 Nov 8;17(Suppl 12):345. doi: 10.1186/s12859-016-1195-2.
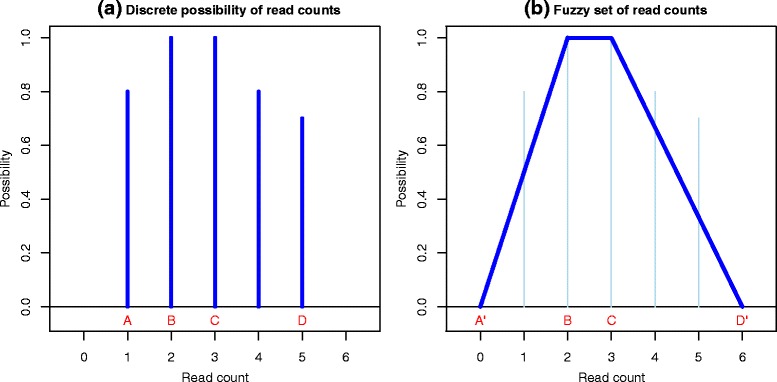
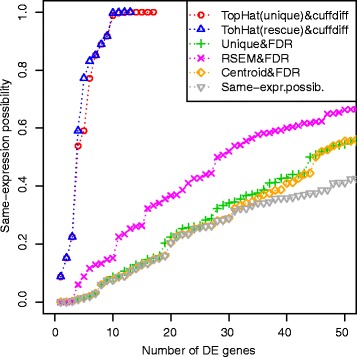
When the reads obtained from high-throughput RNA sequencing are mapped against a reference database, a significant proportion of them - known as multireads - can map to more than one reference sequence. These multireads originate from gene duplications, repetitive regions or overlapping genes. Removing the multireads from the mapping results, in RNA-Seq analyses, causes an underestimation of the read counts, while estimating the real read count can lead to false positives during the detection of differentially expressed sequences.
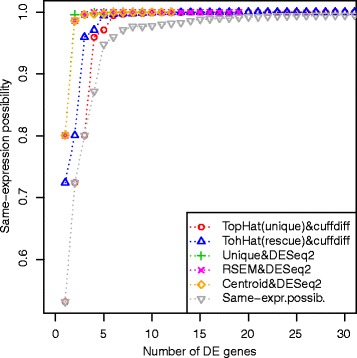
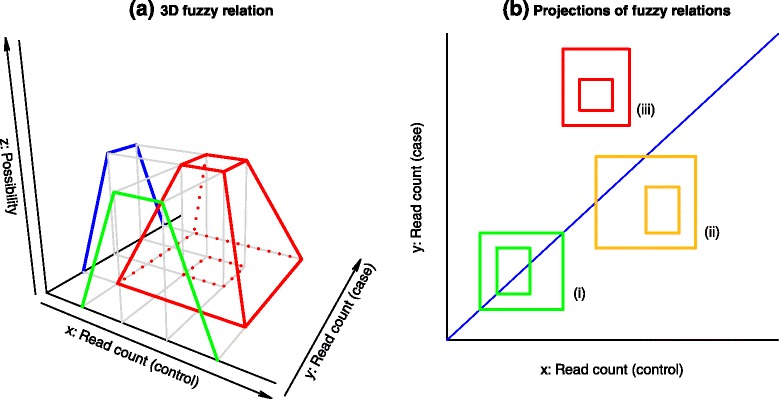
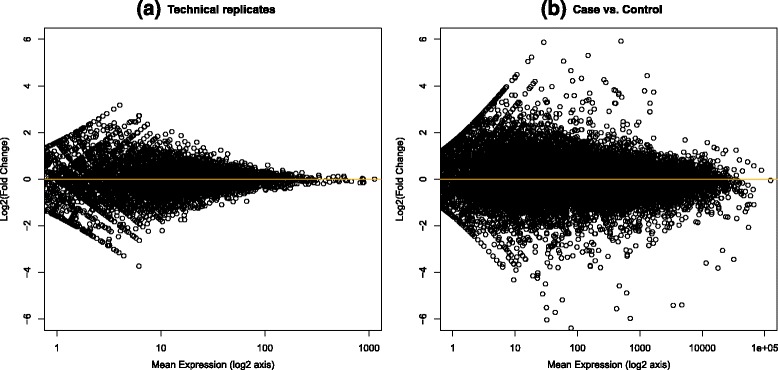
We present an innovative approach to deal with multireads and evaluate differential expression events, entirely based on fuzzy set theory. Since multireads cause uncertainty in the estimation of read counts during gene expression computation, they can also influence the reliability of differential expression analysis results, by producing false positives. Our method manages the uncertainty in gene expression estimation by defining the fuzzy read counts and evaluates the possibility of a gene to be differentially expressed with three fuzzy concepts: over-expression, same-expression and under-expression. The output of the method is a list of differentially expressed genes enriched with information about the uncertainty of the results due to the multiread presence. We have tested the method on RNA-Seq data designed for case-control studies and we have compared the obtained results with other existing tools for read count estimation and differential expression analysis.
The management of multireads with the use of fuzzy sets allows to obtain a list of differential expression events which takes in account the uncertainty in the results caused by the presence of multireads. Such additional information can be used by the biologists when they have to select the most relevant differential expression events to validate with laboratory assays. Our method can be used to compute reliable differential expression events and to highlight possible false positives in the lists of differentially expressed genes computed with other tools.
当将从高通量RNA测序获得的 reads 与参考数据库进行比对时,其中很大一部分——即所谓的多 reads——可以比对到多个参考序列。这些多 reads 源自基因重复、重复区域或重叠基因。在RNA-Seq分析中,从比对结果中去除多 reads 会导致 reads 计数被低估,而估计实际的 reads 计数在检测差异表达序列时可能会导致假阳性。
我们提出了一种创新方法来处理多 reads 并评估差异表达事件,该方法完全基于模糊集理论。由于多 reads 在基因表达计算过程中会导致 reads 计数估计的不确定性,它们还会通过产生假阳性来影响差异表达分析结果的可靠性。我们的方法通过定义模糊 reads 计数来管理基因表达估计中的不确定性,并使用三个模糊概念:过表达、同表达和低表达来评估基因差异表达的可能性。该方法的输出是一个差异表达基因列表,其中丰富了由于多 reads 的存在而导致的结果不确定性信息。我们在为病例对照研究设计的RNA-Seq数据上测试了该方法,并将获得的结果与其他现有的 reads 计数估计和差异表达分析工具进行了比较。
使用模糊集处理多 reads 可以得到一个差异表达事件列表,该列表考虑了由于多 reads 的存在而导致的结果不确定性。当生物学家必须选择最相关的差异表达事件进行实验室检测验证时,这些额外信息可供他们使用。我们的方法可用于计算可靠的差异表达事件,并突出其他工具计算的差异表达基因列表中可能存在的假阳性。