Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Cambridge CB2 0RE, United Kingdom.
Wellcome Trust and MRC Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 0XY, United Kingdom.
Genome Res. 2017 Nov;27(11):1795-1806. doi: 10.1101/gr.222877.117. Epub 2017 Oct 13.
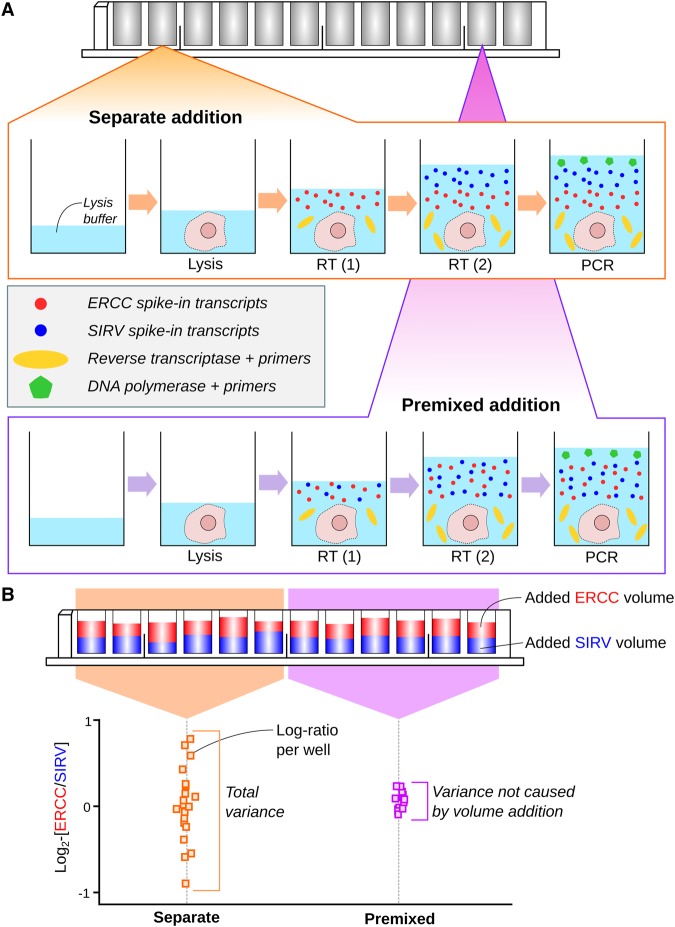
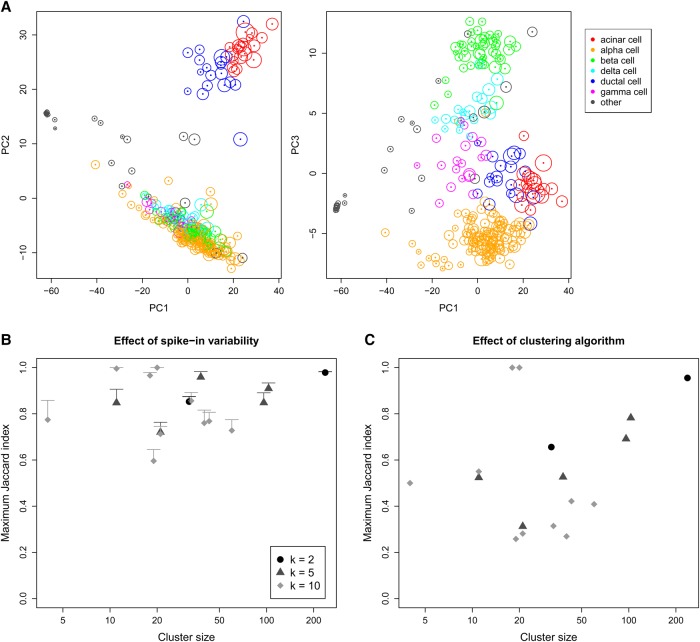
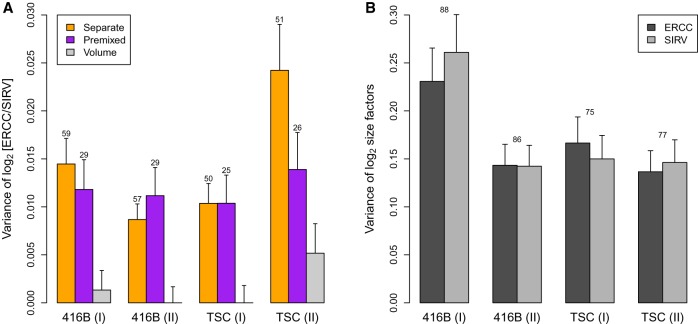
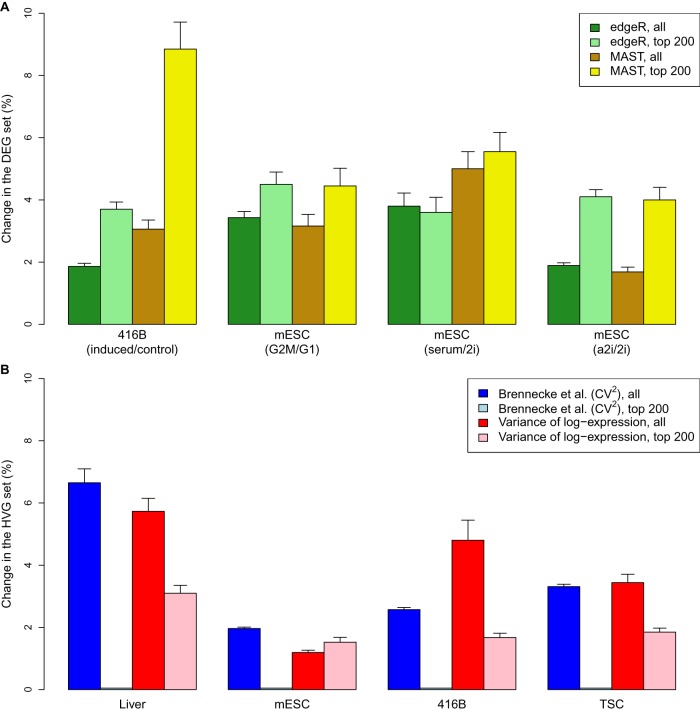
By profiling the transcriptomes of individual cells, single-cell RNA sequencing provides unparalleled resolution to study cellular heterogeneity. However, this comes at the cost of high technical noise, including cell-specific biases in capture efficiency and library generation. One strategy for removing these biases is to add a constant amount of spike-in RNA to each cell and to scale the observed expression values so that the coverage of spike-in transcripts is constant across cells. This approach has previously been criticized as its accuracy depends on the precise addition of spike-in RNA to each sample. Here, we perform mixture experiments using two different sets of spike-in RNA to quantify the variance in the amount of spike-in RNA added to each well in a plate-based protocol. We also obtain an upper bound on the variance due to differences in behavior between the two spike-in sets. We demonstrate that both factors are small contributors to the total technical variance and have only minor effects on downstream analyses, such as detection of highly variable genes and clustering. Our results suggest that scaling normalization using spike-in transcripts is reliable enough for routine use in single-cell RNA sequencing data analyses.
通过对单个细胞的转录组进行分析,单细胞 RNA 测序为研究细胞异质性提供了无与伦比的分辨率。然而,这是以高技术噪声为代价的,包括捕获效率和文库生成方面的细胞特异性偏差。一种消除这些偏差的策略是向每个细胞添加恒定数量的 Spike-in RNA,并对观察到的表达值进行缩放,以使 Spike-in 转录本的覆盖度在细胞间保持恒定。这种方法之前曾受到批评,因为其准确性取决于向每个样本中精确添加 Spike-in RNA。在这里,我们使用两种不同的 Spike-in RNA 进行混合物实验,以量化基于平板方案的每个孔中添加的 Spike-in RNA 的量的方差。我们还获得了由于两个 Spike-in 集之间行为差异引起的方差的上限。我们证明这两个因素都是总技术方差的小贡献者,并且对下游分析(例如高度可变基因的检测和聚类)只有很小的影响。我们的结果表明,使用 Spike-in 转录本进行缩放归一化足以可靠地用于单细胞 RNA 测序数据分析的常规用途。