Department of Pharmacology, Institute of Medical Sciences, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
Experimental Teaching Center of Basic Medicine, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200025, China.
J Neuroinflammation. 2017 Oct 16;14(1):203. doi: 10.1186/s12974-017-0973-8.
Reactive astrogliosis is one of the significantly pathological features in ischemic stroke accompanied with changes in gene expression, morphology, and proliferation. KCa3.1 was involved in TGF-β-induced astrogliosis in vitro and also contributed to astrogliosis-mediated neuroinflammation in neurodegeneration disease.
Wild type mice and KCa3.1 mice were subjected to permanent middle cerebral artery occlusion (pMCAO) to evaluate the infarct areas by 2,3,5-triphenyltetrazolium hydrochloride staining and neurological deficit. KCa3.1 channels expression and cell localization in the brain of pMCAO mice model were measured by immunoblotting and immunostaining. Glia activation and neuron loss was measured by immunostaining. DiBAC4 (3) and Fluo-4AM were used to measure membrane potential and cytosolic Ca level in oxygen-glucose deprivation induced reactive astrocytes in vitro.
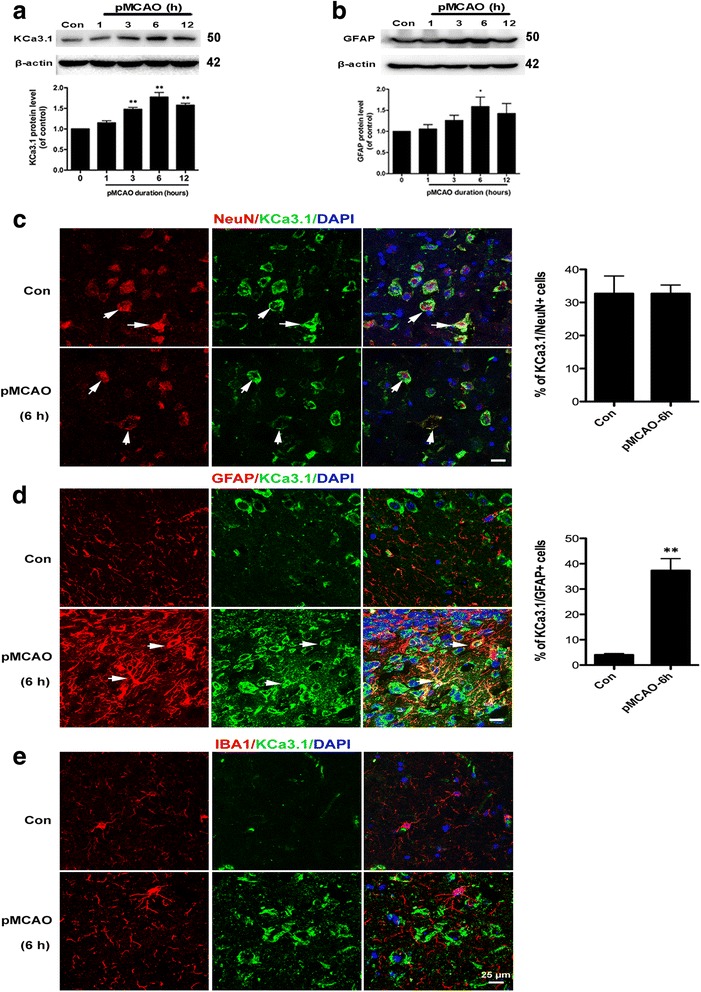
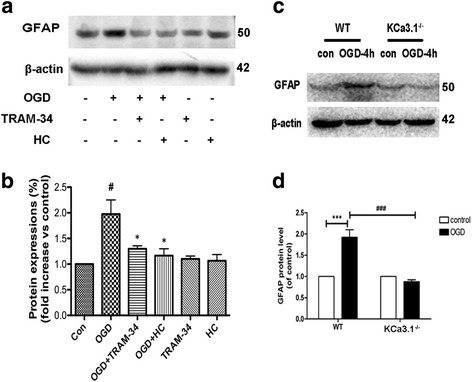
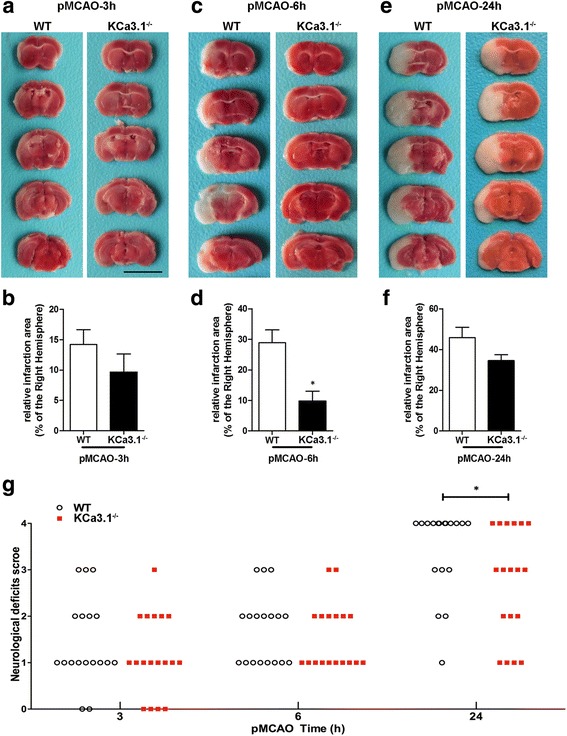
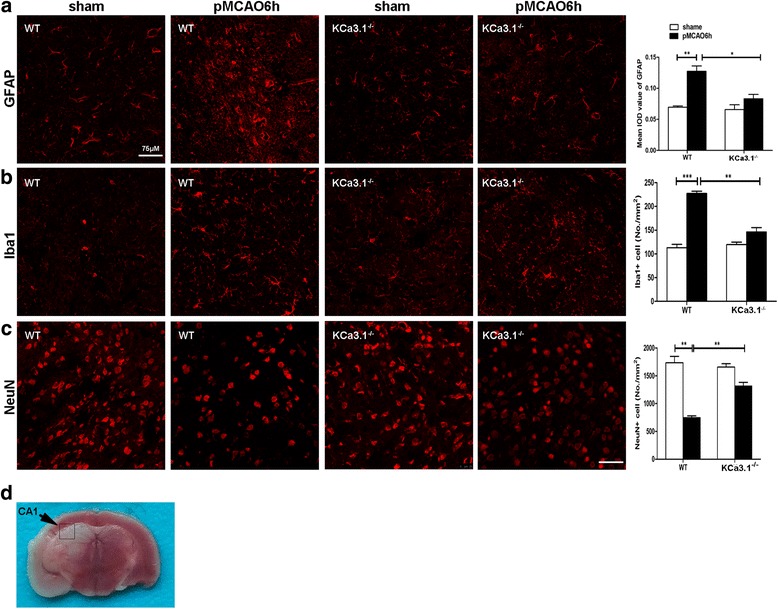
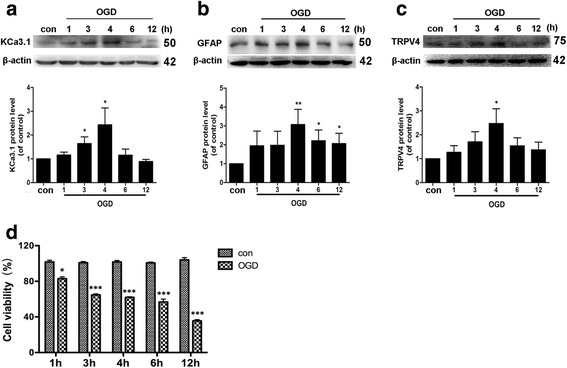
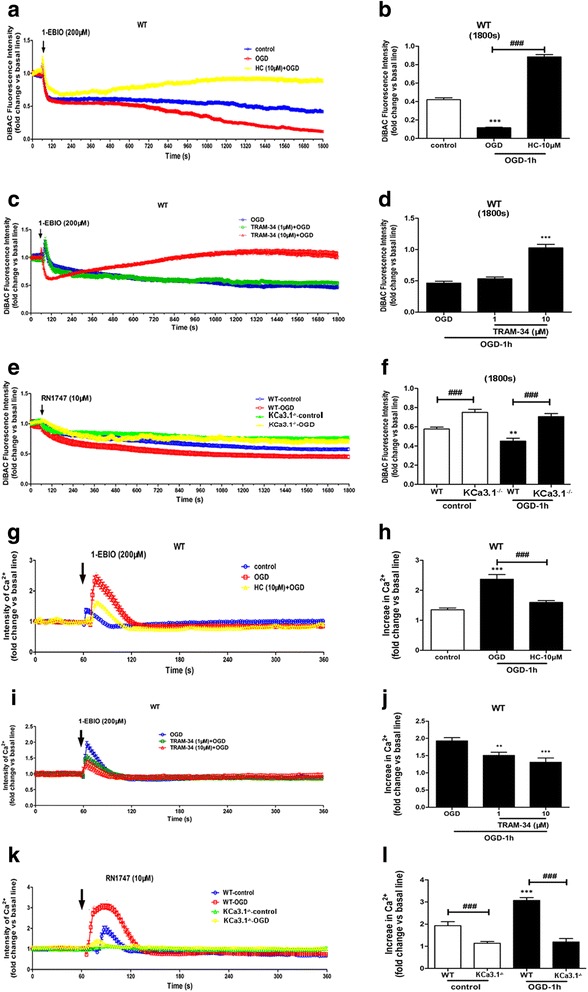
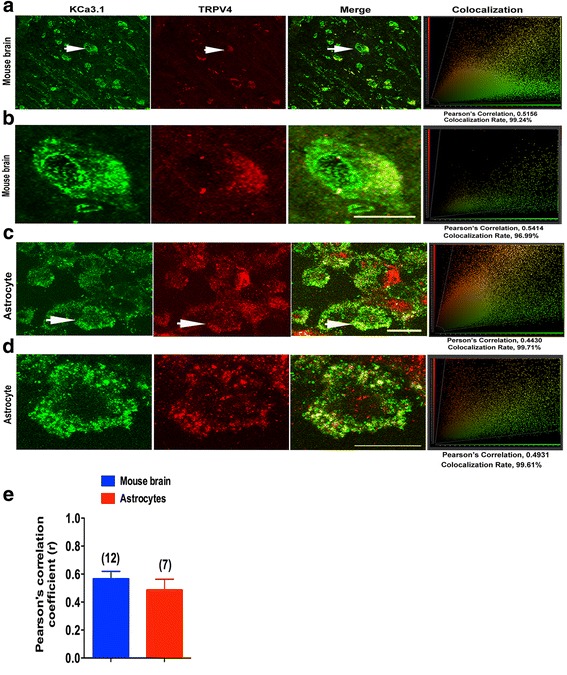
Immunohistochemistry on pMCAO mice infarcts showed strong upregulation of KCa3.1 immunoreactivity in reactive astrogliosis. KCa3.1 mice exhibited significantly smaller infarct areas on pMCAO and improved neurological deficit. Both activated gliosis and neuronal loss were attenuated in KCa3.1 pMCAO mice. In the primary cultured astrocytes, the expressions of KCa3.1 and TRPV4 were increased associated with upregulation of astrogliosis marker GFAP induced by oxygen-glucose deprivation. The activation of KCa3.1 hyperpolarized membrane potential and, by promoting the driving force for calcium, induced calcium entry through TRPV4, a cation channel of the transient receptor potential family. Double-labeled staining showed that KCa3.1 and TRPV4 channels co-localized in astrocytes. Blockade of KCa3.1 or TRPV4 inhibited the phenotype switch of reactive astrogliosis.
Our data suggested that KCa3.1 inhibition might represent a promising therapeutic strategy for ischemia stroke.
反应性星形胶质细胞增生是缺血性中风的显著病理特征之一,伴有基因表达、形态和增殖的变化。KCa3.1 参与了 TGF-β 诱导的体外星形胶质细胞增生,也参与了神经退行性疾病中星形胶质细胞增生介导的神经炎症。
野生型小鼠和 KCa3.1 小鼠接受永久性大脑中动脉闭塞(pMCAO)以通过 2,3,5-三苯基四唑氯化物染色和神经功能缺损评估梗死面积。通过免疫印迹和免疫染色测量 pMCAO 小鼠模型中 KCa3.1 通道的表达和细胞定位。通过免疫染色测量胶质激活和神经元丢失。使用 DiBAC4(3)和 Fluo-4AM 测量体外氧葡萄糖剥夺诱导的反应性星形胶质细胞的膜电位和胞质 Ca 水平。
pMCAO 小鼠梗死的免疫组化显示反应性星形胶质细胞中 KCa3.1 免疫反应性明显上调。KCa3.1 小鼠在 pMCAO 时表现出明显较小的梗死面积和改善的神经功能缺损。KCa3.1 pMCAO 小鼠中的激活胶质增生和神经元丢失均减轻。在原代培养的星形胶质细胞中,KCa3.1 和 TRPV4 的表达增加,与氧葡萄糖剥夺诱导的星形胶质细胞标志物 GFAP 的上调有关。KCa3.1 的激活使膜电位超极化,并通过促进钙的驱动力,通过瞬时受体电位家族的阳离子通道 TRPV4 诱导钙内流。双标记染色显示 KCa3.1 和 TRPV4 通道在星形胶质细胞中共同定位。KCa3.1 或 TRPV4 的阻断抑制了反应性星形胶质细胞的表型转换。
我们的数据表明,KCa3.1 抑制可能代表缺血性中风的一种有前途的治疗策略。