Li Dinghua, Huang Yukun, Leung Chi-Ming, Luo Ruibang, Ting Hing-Fung, Lam Tak-Wah
Department of Computer Science, University of Hong Kong, Pokfulam, Hong Kong.
L3 Bioinformatics Limited, Western District, Hong Kong.
BMC Bioinformatics. 2017 Oct 16;18(Suppl 12):408. doi: 10.1186/s12859-017-1825-3.
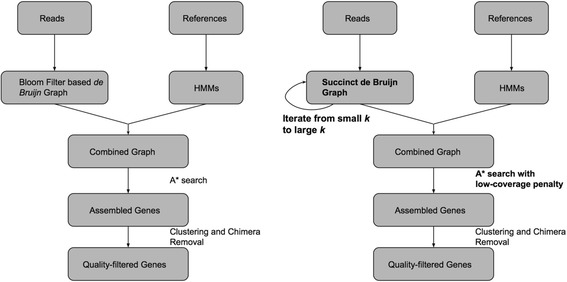
The recent release of the gene-targeted metagenomics assembler Xander has demonstrated that using the trained Hidden Markov Model (HMM) to guide the traversal of de Bruijn graph gives obvious advantage over other assembly methods. Xander, as a pilot study, indeed has a lot of room for improvement. Apart from its slow speed, Xander uses only 1 k-mer size for graph construction and whatever choice of k will compromise either sensitivity or accuracy. Xander uses a Bloom-filter representation of de Bruijn graph to achieve a lower memory footprint. Bloom filters bring in false positives, and it is not clear how this would impact the quality of assembly. Xander does not keep track of the multiplicity of k-mers, which would have been an effective way to differentiate between erroneous k-mers and correct k-mers.
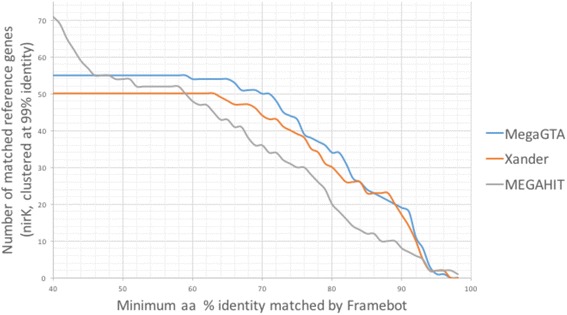
In this paper, we present a new gene-targeted assembler MegaGTA, which attempts to improve Xander in different aspects. Quality-wise, it utilizes iterative de Bruijn graphs to take full advantage of multiple k-mer sizes to make the best of both sensitivity and accuracy. Computation-wise, it employs succinct de Bruijn graphs (SdBG) to achieve low memory footprint and high speed (the latter is benefited from a highly efficient parallel algorithm for constructing SdBG). Unlike Bloom filters, an SdBG is an exact representation of a de Bruijn graph. It enables MegaGTA to avoid false-positive contigs and to easily incorporate the multiplicity of k-mers for building better HMM model. We have compared MegaGTA and Xander on an HMP-defined mock metagenomic dataset, and showed that MegaGTA excelled in both sensitivity and accuracy. On a large rhizosphere soil metagenomic sample (327Gbp), MegaGTA produced 9.7-19.3% more contigs than Xander, and these contigs were assigned to 10-25% more gene references. In our experiments, MegaGTA, depending on the number of k-mers used, is two to ten times faster than Xander.
MegaGTA improves on the algorithm of Xander and achieves higher sensitivity, accuracy and speed. Moreover, it is capable of assembling gene sequences from ultra-large metagenomic datasets. Its source code is freely available at https://github.com/HKU-BAL/megagta .
基因靶向宏基因组组装工具Xander的最新发布表明,使用经过训练的隐马尔可夫模型(HMM)来指导德布鲁因图的遍历比其他组装方法具有明显优势。作为一项初步研究,Xander确实有很大的改进空间。除了速度慢之外,Xander在构建图时仅使用1个k-mer大小,而无论选择何种k值都会在灵敏度或准确性上有所折衷。Xander使用布隆过滤器来表示德布鲁因图以降低内存占用。布隆过滤器会引入误报,并且尚不清楚这将如何影响组装质量。Xander没有跟踪k-mer的多重性,而这本来是区分错误k-mer和正确k-mer的有效方法。
在本文中,我们提出了一种新的基因靶向组装工具MegaGTA,它试图在不同方面改进Xander。在质量方面,它利用迭代德布鲁因图充分利用多个k-mer大小,以兼顾灵敏度和准确性。在计算方面,它采用简洁德布鲁因图(SdBG)来实现低内存占用和高速度(后者受益于用于构建SdBG的高效并行算法)。与布隆过滤器不同,SdBG是德布鲁因图的精确表示。这使得MegaGTA能够避免产生误报重叠群,并能轻松纳入k-mer的多重性以构建更好的HMM模型。我们在一个由人类微生物组计划(HMP)定义的模拟宏基因组数据集上对MegaGTA和Xander进行了比较,结果表明MegaGTA在灵敏度和准确性方面均表现出色。在一个大型根际土壤宏基因组样本(327Gbp)上,MegaGTA产生的重叠群比Xander多9.7 - 19.3%,并且这些重叠群被分配到的基因参考多10 - 25%。在我们的实验中,根据所使用的k-mer数量,MegaGTA比Xander快两到十倍。
MegaGTA改进了Xander的算法,实现了更高的灵敏度、准确性和速度。此外,它能够从超大型宏基因组数据集中组装基因序列。其源代码可在https://github.com/HKU - BAL/megagta免费获取。