Computer Science and Engineering, Michigan State Univerisity, 428 South Shaw Rd East Lansing, MI 48824, USA and Center for Microbial Ecology, Michigan State University, East Lansing, MI 48824, USA.
Bioinformatics. 2015 Jun 15;31(12):i35-43. doi: 10.1093/bioinformatics/btv231.
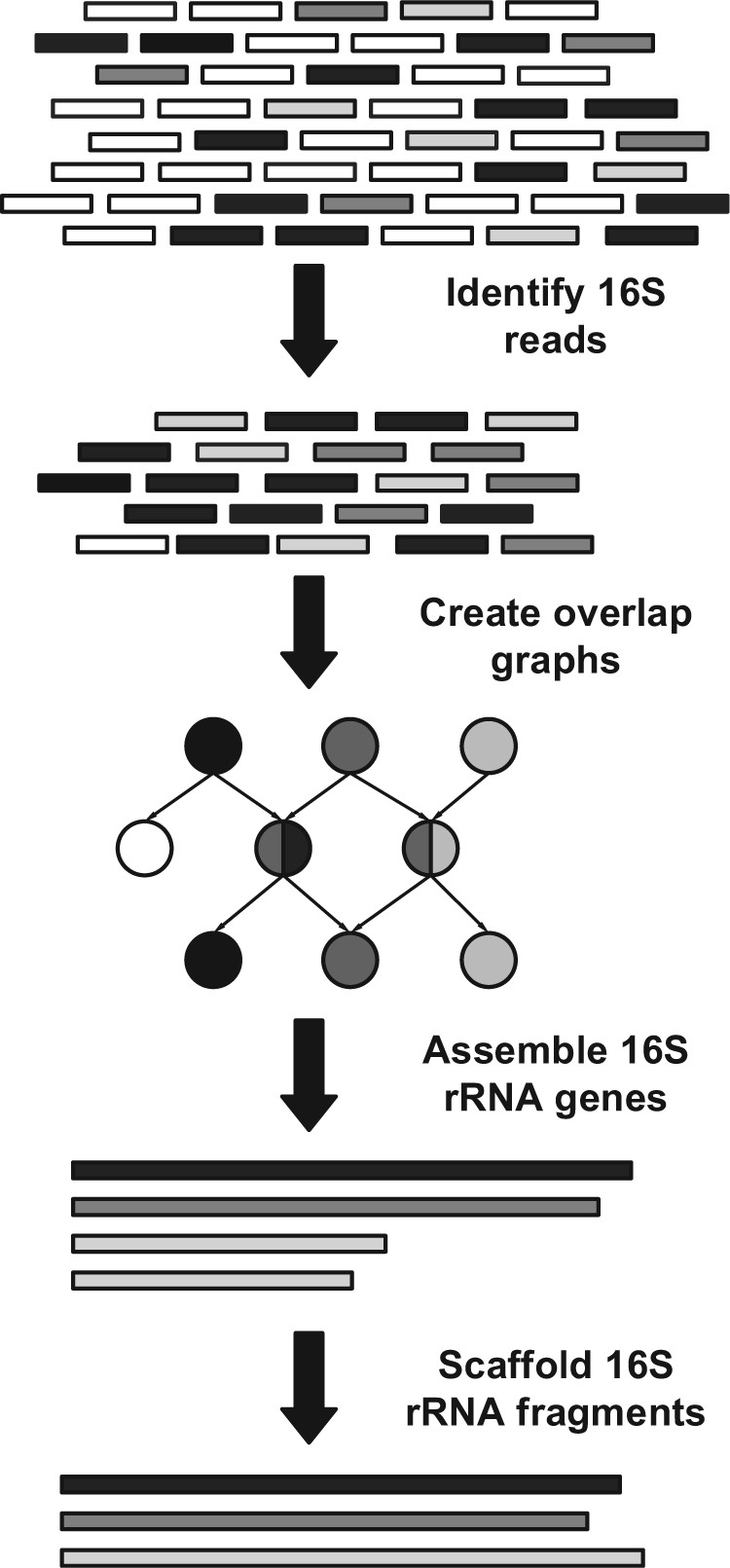
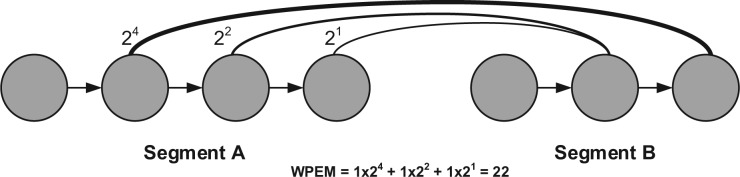
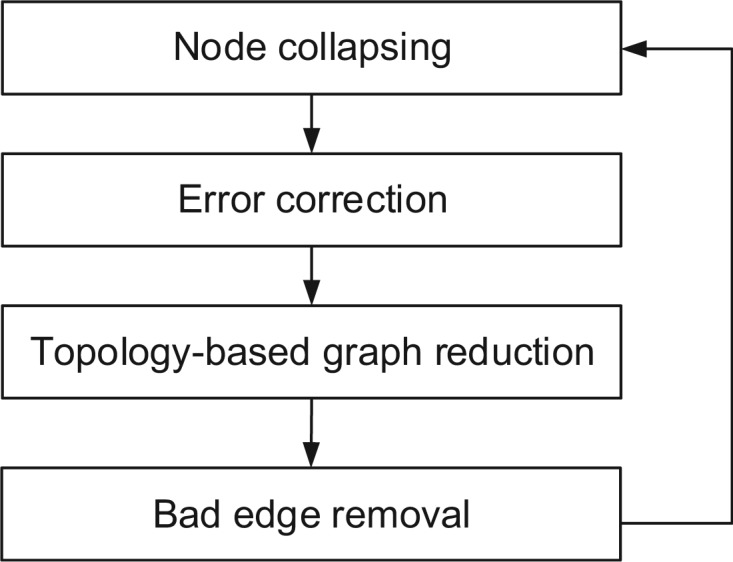
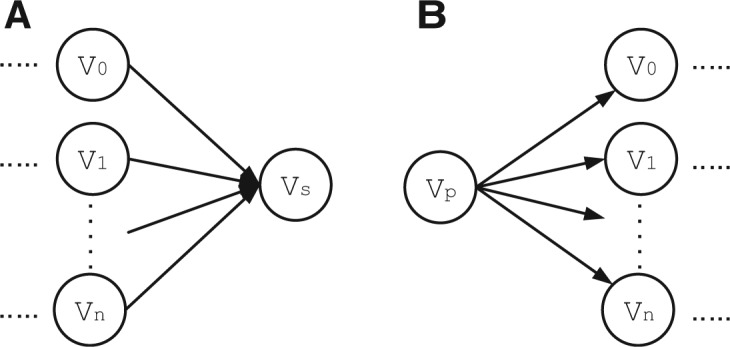
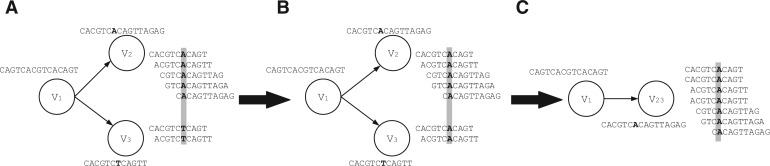
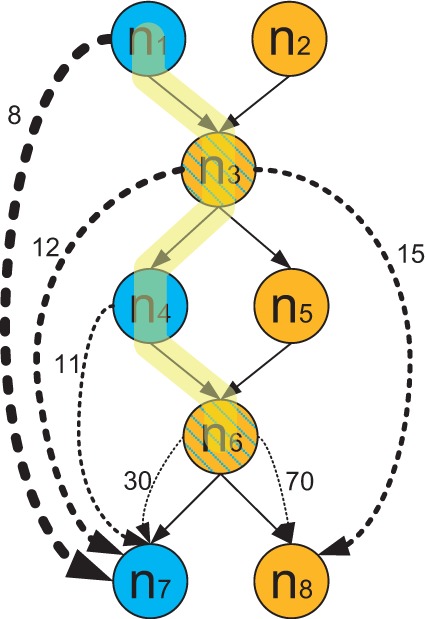
Metagenomic data, which contains sequenced DNA reads of uncultured microbial species from environmental samples, provide a unique opportunity to thoroughly analyze microbial species that have never been identified before. Reconstructing 16S ribosomal RNA, a phylogenetic marker gene, is usually required to analyze the composition of the metagenomic data. However, massive volume of dataset, high sequence similarity between related species, skewed microbial abundance and lack of reference genes make 16S rRNA reconstruction difficult. Generic de novo assembly tools are not optimized for assembling 16S rRNA genes. In this work, we introduce a targeted rRNA assembly tool, REAGO (REconstruct 16S ribosomal RNA Genes from metagenOmic data). It addresses the above challenges by combining secondary structure-aware homology search, zproperties of rRNA genes and de novo assembly. Our experimental results show that our tool can correctly recover more rRNA genes than several popular generic metagenomic assembly tools and specially designed rRNA construction tools.
The source code of REAGO is freely available at https://github.com/chengyuan/reago.
宏基因组数据包含了从环境样本中未培养微生物物种的测序 DNA 片段,为彻底分析以前从未鉴定过的微生物物种提供了独特的机会。通常需要重建 16S 核糖体 RNA(一种系统发育标记基因)来分析宏基因组数据的组成。然而,数据集的巨大体积、相关物种之间的高序列相似性、微生物丰度的倾斜以及缺乏参考基因使得 16S rRNA 的重建变得困难。通用的从头组装工具不能针对 16S rRNA 基因进行优化。在这项工作中,我们引入了一种靶向 rRNA 组装工具,REAGO(从宏基因组数据中重建 16S 核糖体 RNA 基因)。它通过结合二级结构感知同源搜索、rRNA 基因的 z 属性和从头组装来解决上述挑战。我们的实验结果表明,与几个流行的通用宏基因组组装工具和专门设计的 rRNA 构建工具相比,我们的工具可以更准确地恢复更多的 rRNA 基因。
REAGO 的源代码可在 https://github.com/chengyuan/reago 上免费获取。