Division of Neurobiology, The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Edinburgh, UK.
Euan MacDonald Centre for Motor Neurone Disease Research, University of Edinburgh, Edinburgh, UK.
Mol Neurodegener. 2017 Oct 27;12(1):77. doi: 10.1186/s13024-017-0221-9.
Neurons are highly polarized cells consisting of three distinct functional domains: the cell body (and associated dendrites), the axon and the synapse. Previously, it was believed that the clinical phenotypes of neurodegenerative diseases were caused by the loss of entire neurons, however it has recently become apparent that these neuronal sub-compartments can degenerate independently, with synapses being particularly vulnerable to a broad range of stimuli. Whilst the properties governing the differential degenerative mechanisms remain unknown, mitochondria consistently appear in the literature, suggesting these somewhat promiscuous organelles may play a role in affecting synaptic stability. Synaptic and non-synaptic mitochondrial subpools are known to have different enzymatic properties (first demonstrated by Lai et al., 1977). However, the molecular basis underpinning these alterations, and their effects on morphology, has not been well documented.
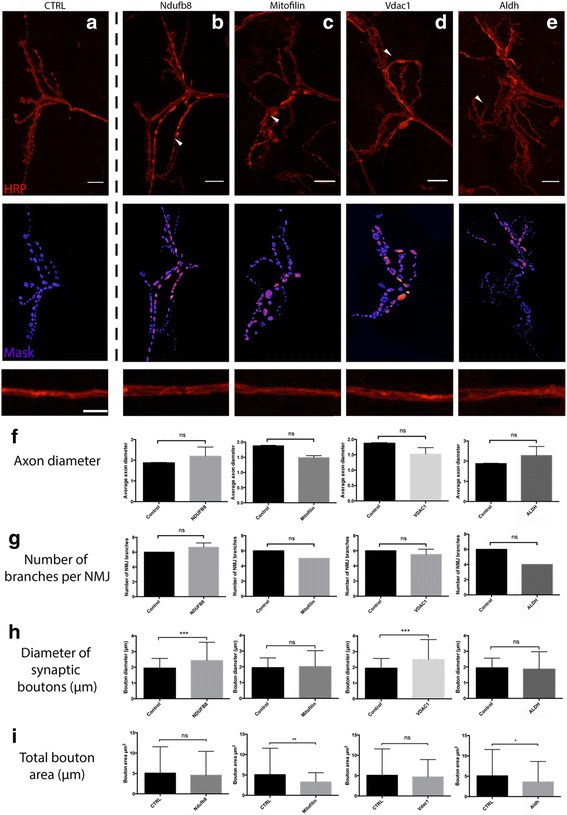
The current study has employed electron microscopy, label-free proteomics and in silico analyses to characterize the morphological and biochemical properties of discrete sub-populations of mitochondria. The physiological relevance of these findings was confirmed in-vivo using a molecular genetic approach at the Drosophila neuromuscular junction.
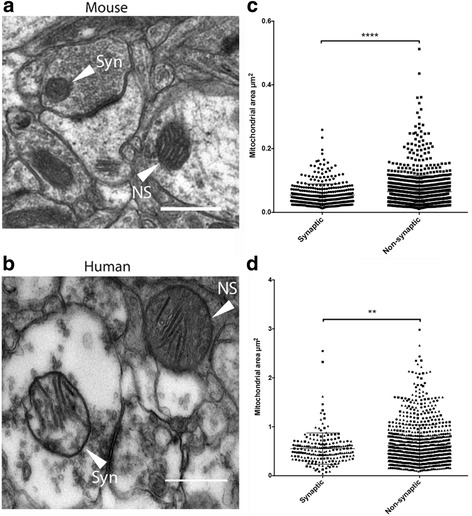
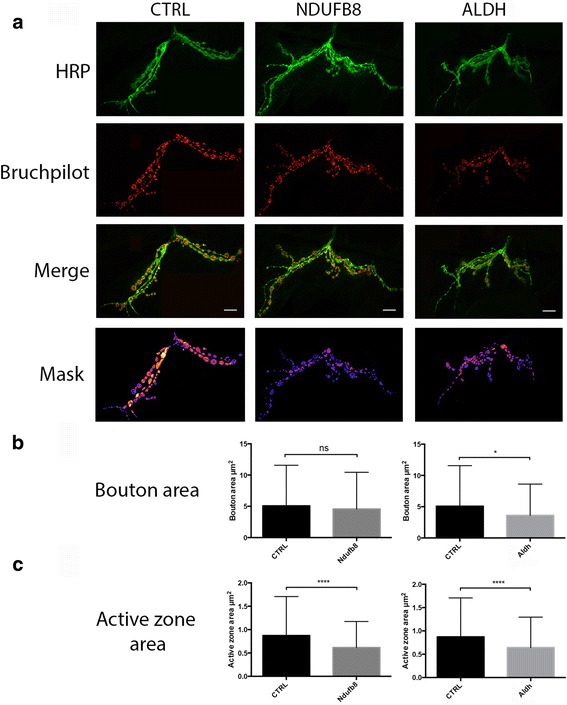
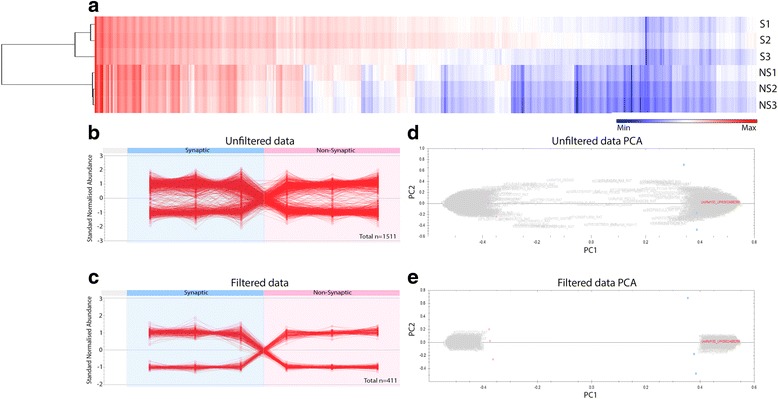
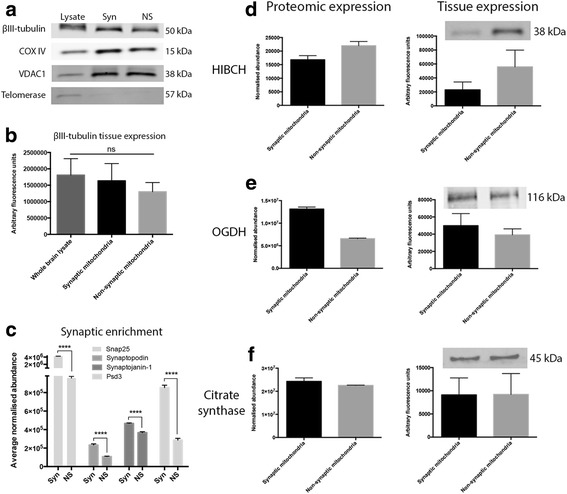
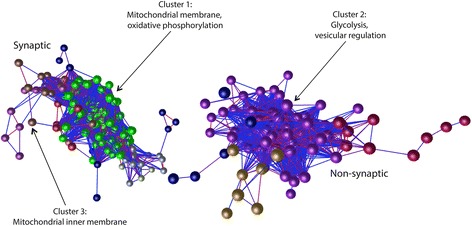
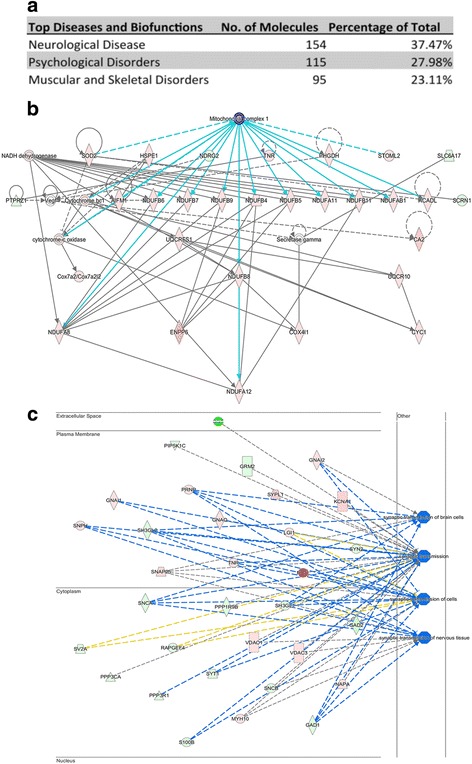
Here, we demonstrate that mitochondria at the synaptic terminal are indeed morphologically different to non-synaptic mitochondria, in both rodents and human patients. Furthermore, generation of proteomic profiles reveals distinct molecular fingerprints - highlighting that the properties of complex I may represent an important specialisation of synaptic mitochondria. Evidence also suggests that at least 30% of the mitochondrial enzymatic activity differences previously reported can be accounted for by protein abundance. Finally, we demonstrate that the molecular differences between discrete mitochondrial sub-populations are capable of selectively influencing synaptic morphology in-vivo. We offer several novel mitochondrial candidates that have the propensity to significantly alter the synaptic architecture in-vivo.
Our study demonstrates discrete proteomic profiles exist dependent upon mitochondrial subcellular localization and selective alteration of intrinsic mitochondrial proteins alters synaptic morphology in-vivo.
神经元是具有三个不同功能域的高度极化细胞:细胞体(和相关树突)、轴突和突触。以前,人们认为神经退行性疾病的临床表型是由整个神经元的丧失引起的,但最近显然这些神经元亚区可以独立退化,其中突触特别容易受到广泛刺激的影响。虽然控制差异退化机制的特性尚不清楚,但线粒体在文献中经常出现,这表明这些有些混杂的细胞器可能在影响突触稳定性方面发挥作用。已知突触和非突触线粒体亚池具有不同的酶特性(首次由 Lai 等人于 1977 年证明)。然而,这些变化的分子基础及其对形态的影响尚未得到很好的记录。
本研究采用电子显微镜、无标记蛋白质组学和计算机分析来表征离散线粒体亚群的形态和生化特性。这些发现的生理相关性在果蝇神经肌肉接头处使用分子遗传方法在体内得到了证实。
在这里,我们证明了在啮齿动物和人类患者中,突触末端的线粒体在形态上确实与非突触线粒体不同。此外,生成蛋白质组学图谱揭示了不同的分子指纹-这表明复合物 I 的特性可能代表突触线粒体的一个重要特化。证据还表明,以前报道的线粒体酶活性差异的至少 30%可以通过蛋白质丰度来解释。最后,我们证明了离散线粒体亚群之间的分子差异能够选择性地影响体内突触形态。我们提供了几个新的线粒体候选物,它们有潜力在体内显著改变突触结构。
我们的研究表明,存在依赖于线粒体亚细胞定位的离散蛋白质组学图谱,并且内在线粒体蛋白的选择性改变会改变体内的突触形态。