Shah Samir A, Ganesan Sukirth M, Varadharaj Saradhadevi, Dabdoub Shareef M, Walters John D, Kumar Purnima S
Division of Periodontology, College of Dentistry, The Ohio State University, 4111 Postle Hall. 305, W 12th Avenue, Columbus, OH 43210 USA.
Present Address: South Jersey Periodontics, New Jersey, USA.
NPJ Biofilms Microbiomes. 2017 Oct 24;3:26. doi: 10.1038/s41522-017-0033-2. eCollection 2017.
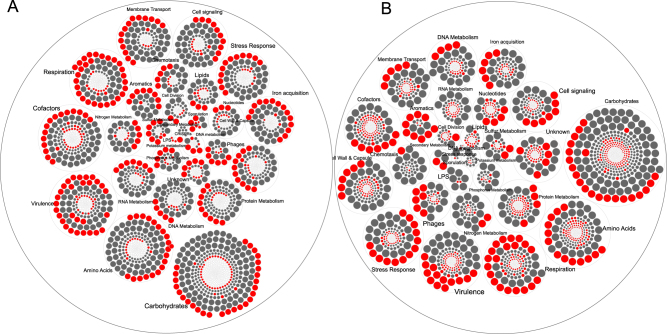
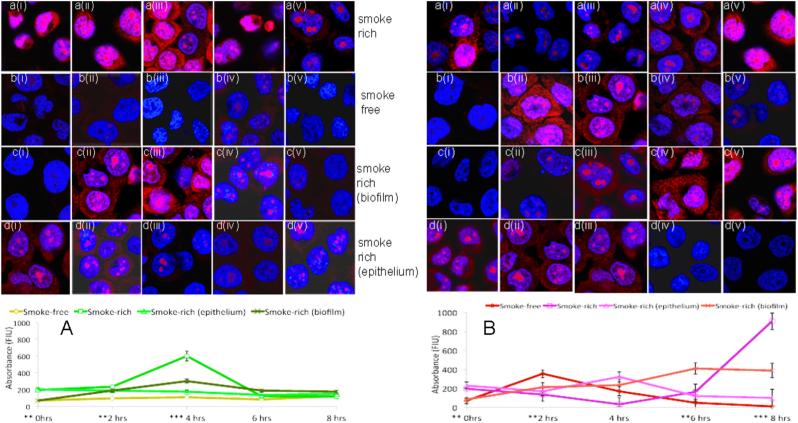
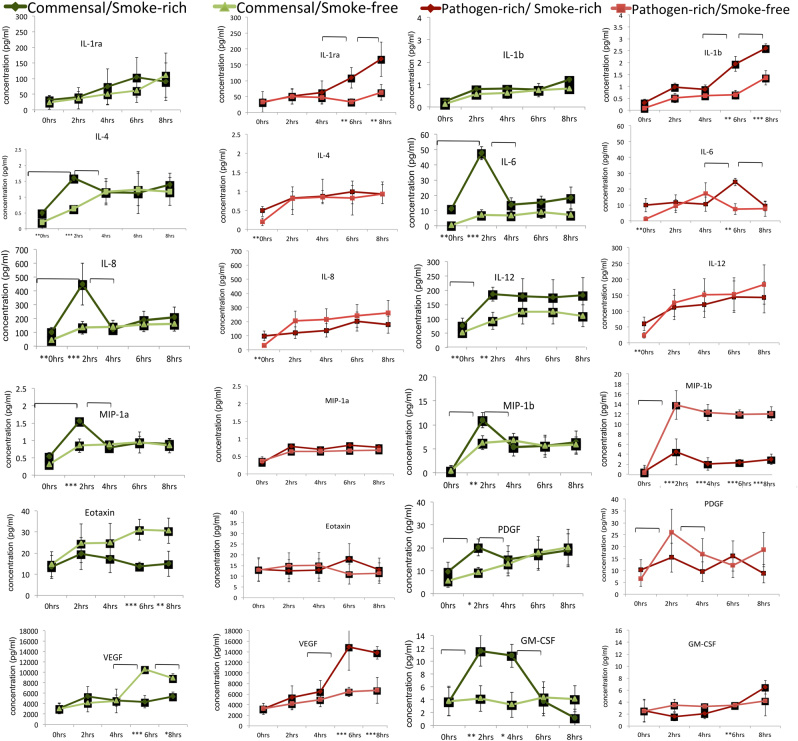
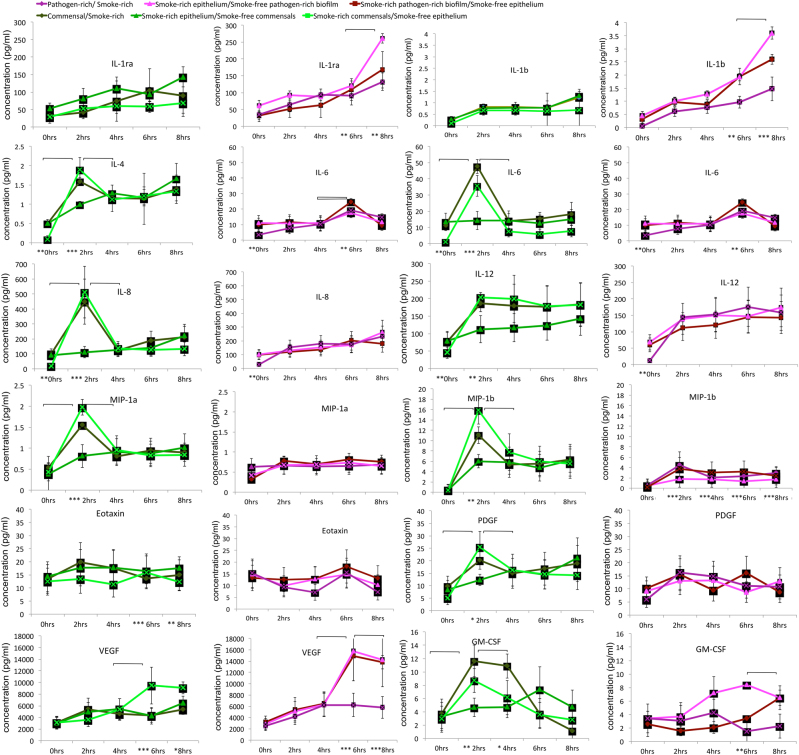
We have previously reported that oral biofilms in clinically healthy smokers are pathogen-rich, and that this enrichment occurs within 24 h of biofilm formation. The present investigation aimed to identify a mechanism by which smoking creates this altered community structure. By combining in vitro microbial-mucosal interface models of commensal (consisting of and and pathogen-rich (comprising and , and communities with metatranscriptomics, targeted proteomics and fluorescent microscopy, we demonstrate that smoke exposure significantly downregulates essential metabolic functions within commensal biofilms, while significantly increasing expression of virulence genes, notably lipopolysaccharide (LPS), flagella and capsule synthesis. By contrast, in pathogen-rich biofilms several metabolic pathways were over-expressed in response to smoke exposure. Under smoke-rich conditions, epithelial cells mounted an early and amplified pro-inflammatory and oxidative stress response to these virulence-enhanced commensal biofilms, and a muted early response to pathogen-rich biofilms. Commensal biofilms also demonstrated early and widespread cell death. Similar results were observed when smoke-free epithelial cells were challenged with smoke-conditioned biofilms, but not vice versa. In conclusion, our data suggest that smoke-induced transcriptional shifts in commensal biofilms triggers a florid pro-inflammatory response, leading to early commensal death, which may preclude niche saturation by these beneficial organisms. The cytokine-rich, pro-oxidant, anaerobic environment sustains inflammophilic bacteria, and, in the absence of commensal antagonism, may promote the creation of pathogen-rich biofilms in smokers.
我们之前曾报道,临床健康吸烟者的口腔生物膜富含病原体,且这种富集在生物膜形成后的24小时内就会发生。本研究旨在确定吸烟导致这种群落结构改变的机制。通过将共生(由 和 组成)和富含病原体(由 、 、 和 组成)的体外微生物 - 黏膜界面模型与宏转录组学、靶向蛋白质组学和荧光显微镜相结合,我们证明,烟雾暴露显著下调了共生生物膜内的基本代谢功能,同时显著增加了毒力基因的表达,尤其是脂多糖(LPS)、鞭毛和荚膜合成相关基因的表达。相比之下,在富含病原体的生物膜中,有几种代谢途径在烟雾暴露后过度表达。在烟雾丰富的条件下,上皮细胞对这些毒力增强的共生生物膜产生了早期且增强的促炎和氧化应激反应,而对富含病原体的生物膜的早期反应则较为微弱。共生生物膜还出现了早期且广泛的细胞死亡。当用经烟雾处理的生物膜挑战无烟上皮细胞时,也观察到了类似结果,但反之则不然。总之,我们的数据表明,烟雾诱导的共生生物膜转录变化引发了强烈的促炎反应,导致早期共生生物死亡,这可能使这些有益微生物无法实现生态位饱和。富含细胞因子、促氧化剂的厌氧环境维持了嗜炎细菌的生长,并且在没有共生拮抗作用的情况下,可能会促进吸烟者中富含病原体的生物膜的形成。