Antunes Dinler A, Moll Mark, Devaurs Didier, Jackson Kyle R, Lizée Gregory, Kavraki Lydia E
Department of Computer Science, Rice University, Houston, Texas.
Department of Melanoma Medical Oncology - Research, The University of Texas MD Anderson Cancer Center, Houston, Texas.
Cancer Res. 2017 Nov 1;77(21):e55-e57. doi: 10.1158/0008-5472.CAN-17-0511.
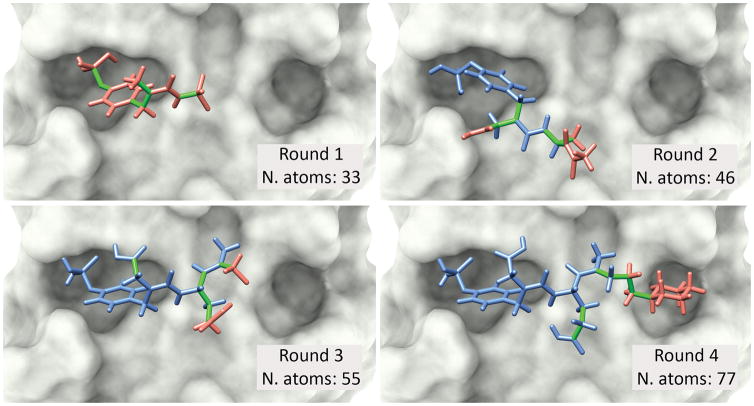
Molecular docking is a standard computational approach to predict binding modes of protein-ligand complexes by exploring alternative orientations and conformations of the ligand (i.e., by exploring ligand flexibility). Docking tools are largely used for virtual screening of small drug-like molecules, but their accuracy and efficiency greatly decays for ligands with more than 10 flexible bonds. This prevents a broader use of these tools to dock larger ligands, such as peptides, which are molecules of growing interest in cancer research. To overcome this limitation, our group has previously proposed a meta-docking strategy, called DINC, to predict binding modes of large ligands. By incrementally docking overlapping fragments of a ligand, DINC allowed predicting binding modes of peptide-based inhibitors of transcription factors involved in cancer. Here, we describe DINC 2.0, a revamped version of the DINC webserver with enhanced capabilities and a more user-friendly interface. DINC 2.0 allows docking ligands that were previously too challenging for DINC, such as peptides with more than 25 flexible bonds. The webserver is freely accessible at http://dinc.kavrakilab.org, together with additional documentation and video tutorials. Our team will provide continuous support for this tool and is working on extending its applicability to other challenging fields, such as personalized immunotherapy against cancer. .
分子对接是一种标准的计算方法,通过探索配体的替代取向和构象(即通过探索配体的灵活性)来预测蛋白质-配体复合物的结合模式。对接工具主要用于小分子药物样分子的虚拟筛选,但对于具有超过10个柔性键的配体,其准确性和效率会大幅下降。这阻碍了这些工具更广泛地用于对接更大的配体,如肽,而肽是癌症研究中越来越受关注的分子。为了克服这一限制,我们团队此前提出了一种称为DINC的元对接策略,以预测大配体的结合模式。通过逐步对接配体的重叠片段,DINC能够预测参与癌症的转录因子的基于肽的抑制剂的结合模式。在此,我们描述了DINC 2.0,这是DINC网络服务器的改进版本,具有增强的功能和更用户友好的界面。DINC 2.0能够对接以前对DINC来说具有挑战性的配体,例如具有超过25个柔性键的肽。该网络服务器可通过http://dinc.kavrakilab.org免费访问,同时还提供了额外的文档和视频教程。我们的团队将为该工具提供持续支持,并正在努力将其适用性扩展到其他具有挑战性的领域,如针对癌症的个性化免疫疗法。