Department of Computer Science, Rice University, 6100 Main St, Houston, TX 77005, USA.
Department of Melanoma Medical Oncology - Research, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030, USA.
BMC Mol Cell Biol. 2019 Sep 5;20(1):42. doi: 10.1186/s12860-019-0218-z.
Docking large ligands, and especially peptides, to protein receptors is still considered a challenge in computational structural biology. Besides the issue of accurately scoring the binding modes of a protein-ligand complex produced by a molecular docking tool, the conformational sampling of a large ligand is also often considered a challenge because of its underlying combinatorial complexity. In this study, we evaluate the impact of using parallelized and incremental paradigms on the accuracy and performance of conformational sampling when docking large ligands. We use five datasets of protein-ligand complexes involving ligands that could not be accurately docked by classical protein-ligand docking tools in previous similar studies.
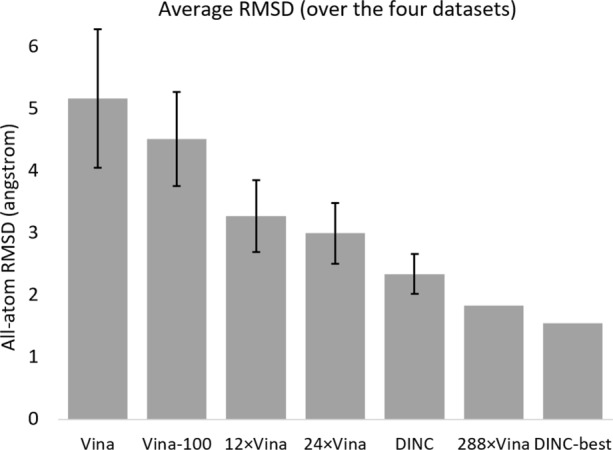
Our computational evaluation shows that simply increasing the amount of conformational sampling performed by a protein-ligand docking tool, such as Vina, by running it for longer is rarely beneficial. Instead, it is more efficient and advantageous to run several short instances of this docking tool in parallel and group their results together, in a straightforward parallelized docking protocol. Even greater accuracy and efficiency are achieved by our parallelized incremental meta-docking tool, DINC, showing the additional benefits of its incremental paradigm. Using DINC, we could accurately reproduce the vast majority of the protein-ligand complexes we considered.
Our study suggests that, even when trying to dock large ligands to proteins, the conformational sampling of the ligand should no longer be considered an issue, as simple docking protocols using existing tools can solve it. Therefore, scoring should currently be regarded as the biggest unmet challenge in molecular docking.
对接大型配体,尤其是肽,仍然被认为是计算结构生物学中的一个挑战。除了准确评分由分子对接工具产生的蛋白质-配体复合物的结合模式的问题外,由于其潜在的组合复杂性,大型配体的构象采样也常常被认为是一个挑战。在这项研究中,我们评估了在对接大型配体时使用并行和增量范式对接对构象采样的准确性和性能的影响。我们使用了五个涉及配体的蛋白质-配体复合物数据集,这些配体在以前的类似研究中无法被经典的蛋白质-配体对接工具准确对接。
我们的计算评估表明,简单地通过运行更长时间来增加蛋白质-配体对接工具(如 Vina)执行的构象采样量很少是有益的。相反,更有效和有利的方法是并行运行几个这种对接工具的短实例,并将它们的结果分组在一起,采用一种简单的并行对接协议。我们的并行增量元对接工具 DINC 实现了更高的准确性和效率,展示了其增量范式的额外优势。使用 DINC,我们可以准确地重现我们考虑的绝大多数蛋白质-配体复合物。
我们的研究表明,即使在尝试对接大型配体到蛋白质时,配体的构象采样也不再被认为是一个问题,因为使用现有工具的简单对接方案可以解决这个问题。因此,评分目前应该被视为分子对接中最大的未满足的挑战。