Department of Biosciences and Informatics, Keio University, Japan.
Brief Bioinform. 2019 May 21;20(3):866-876. doi: 10.1093/bib/bbx147.
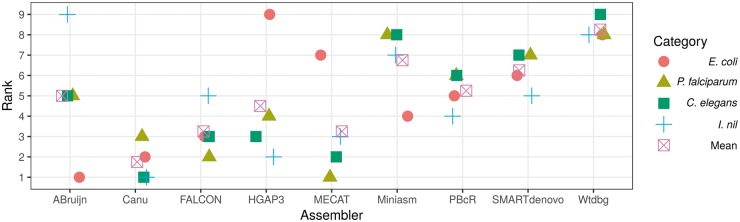
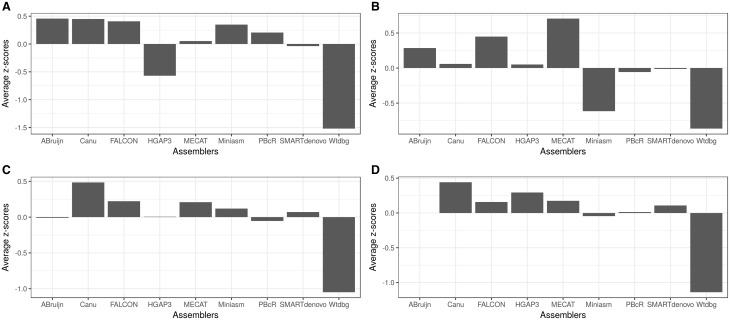
Long reads obtained from third-generation sequencing platforms can help overcome the long-standing challenge of the de novo assembly of sequences for the genomic analysis of non-model eukaryotic organisms. Numerous long-read-aided de novo assemblies have been published recently, which exhibited superior quality of the assembled genomes in comparison with those achieved using earlier second-generation sequencing technologies. Evaluating assemblies is important in guiding the appropriate choice for specific research needs. In this study, we evaluated 10 long-read assemblers using a variety of metrics on Pacific Biosciences (PacBio) data sets from different taxonomic categories with considerable differences in genome size. The results allowed us to narrow down the list to a few assemblers that can be effectively applied to eukaryotic assembly projects. Moreover, we highlight how best to use limited genomic resources for effectively evaluating the genome assemblies of non-model organisms.
第三代测序平台获得的长读长有助于克服非模式真核生物基因组分析中从头组装序列的长期存在的挑战。最近发表了许多基于长读长的从头组装,与使用早期第二代测序技术相比,组装的基因组质量更高。评估组装对于指导针对特定研究需求的适当选择很重要。在这项研究中,我们使用多种指标在不同分类群的太平洋生物科学(PacBio)数据集上评估了 10 种长读长组装器,这些数据集在基因组大小方面存在相当大的差异。结果使我们能够将列表缩小到少数几个可以有效地应用于真核生物组装项目的组装器。此外,我们还强调了如何有效地利用有限的基因组资源来有效地评估非模式生物的基因组组装。