Zhang Tianguang, Ikejima Takashi, Li Lizhong, Wu Ruiqin, Yuan Xiaoyan, Zhao Jun, Wang Yimei, Peng Shuangqing
Evaluation and Research Center for Toxicology, Institute of Disease Control and Prevention, PLA, Beijing, China.
China-Japan Research Institute of Medical and Pharmaceutical Sciences, Shenyang Pharmaceutical University, Shenyang, China.
Front Pharmacol. 2017 Oct 26;8:753. doi: 10.3389/fphar.2017.00753. eCollection 2017.
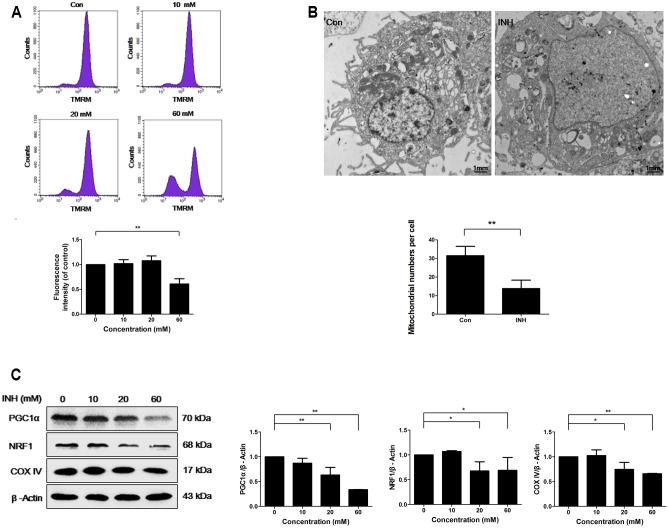
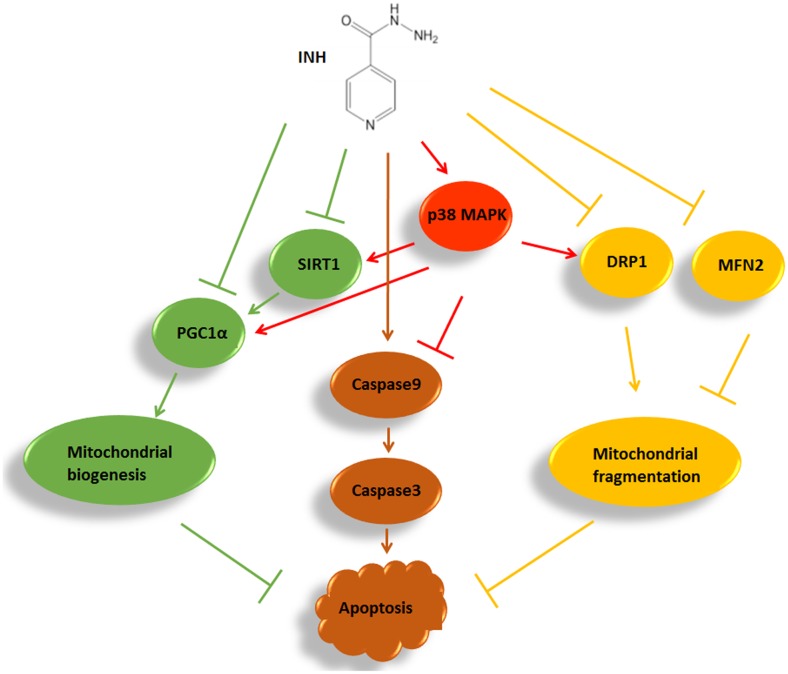
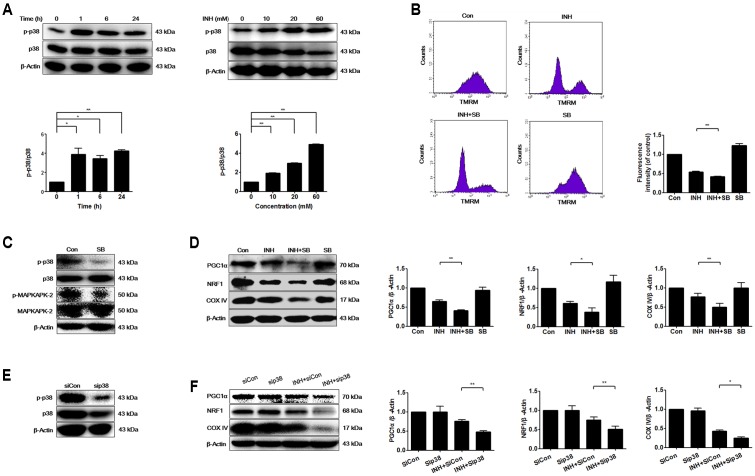
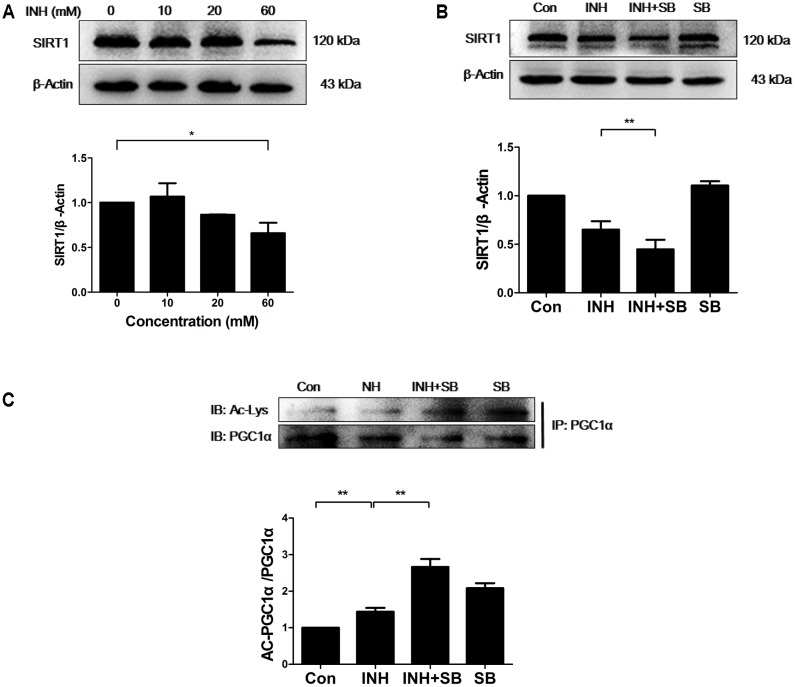
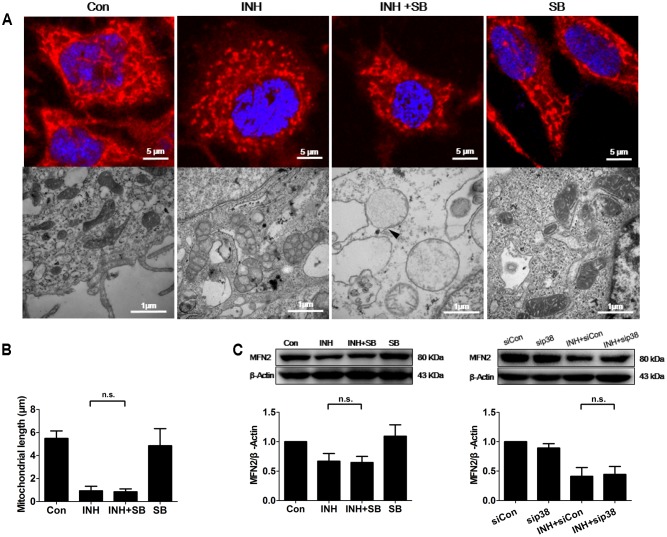
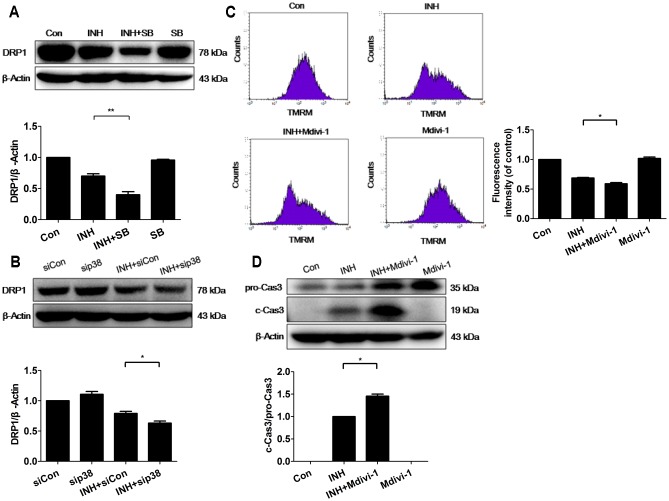
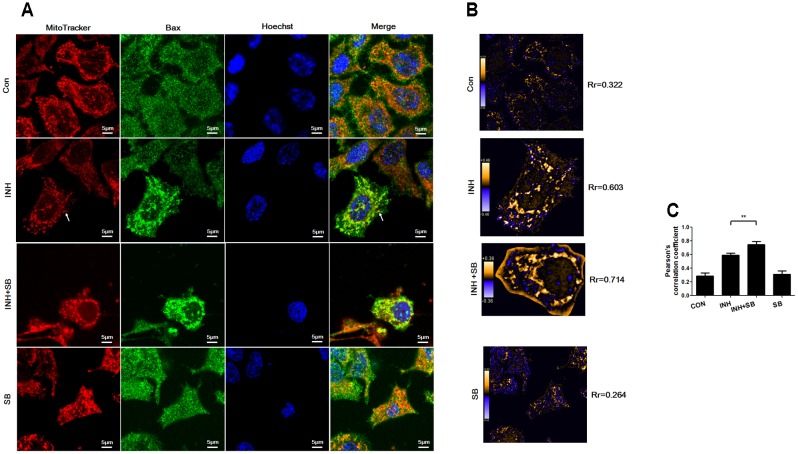
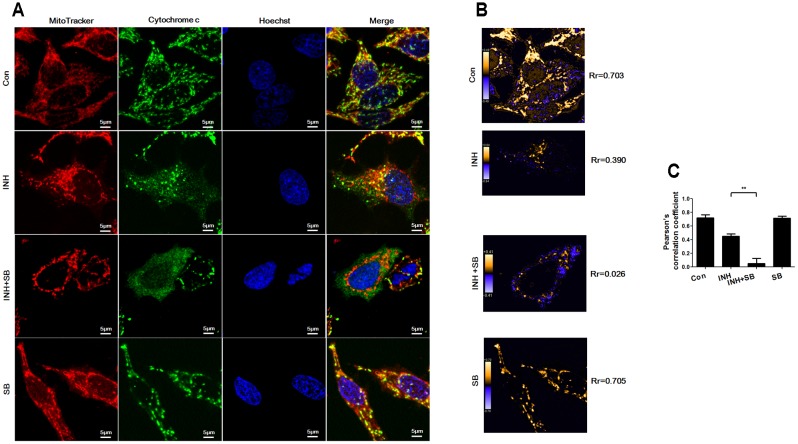
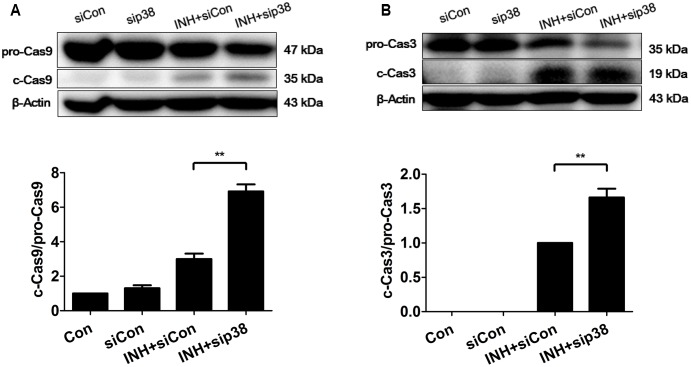
Isoniazid (INH), a widely used first-line antitubercular drug, has been noted to be associated with hepatotoxicity. In spite of extensive researches over many decades, the mechanism of INH-induced hepatotoxicity still remains poorly understood. Recently, mitochondrial toxicity has been emerging as a new paradigm for INH-induced hepatotoxicity. In this study, we showed that INH impaired mitochondrial biogenesis and dynamics in human hepatocarcinoma HepG2 cells. INH reduced mitochondrial membrane potential (MMP) and induced mitochondria swelling. INH also inhibited the protein expressions of three major mitochondrial biogenesis regulators, SIRT1, PGC1α and NRF1, along with increased acetylation of PGC1α. Meanwhile, INH decreased the number of mitochondria, accompanied by decreased expression of mitochondrial protein COX IV. INH caused mitochondrial fragmentation involving decreased levels of the fusion protein MFN2 as well as the fission protein DRP1. INH-reduced DRP1 expression was associated with the increase of apoptosis, suggesting the existence of pro-survival fission and its involvement in mitochondrial quality control. INH activated p38 MAPK, whereas inhibition of p38 MAPK aggravated INH-induced decreases of SIRT1, PGC1α, NRF1, COX IV and DRP1 expressions. P38 MAPK inhibition also further up-regulated the acetylation of PGC1α and exacerbated INH-induced MMP loss, mitochondrial swelling and apoptosis. Taken together, INH-activated p38 MAPK induced mitochondrial biogenesis to alleviate apoptosis through partly recovering SIRT1-PGC1α pathway activation. In the meantime, p38 MAPK activation by INH promoted protective mitochondrial fission to alleviate apoptosis by partial recovery of DRP1 expression.
异烟肼(INH)是一种广泛使用的一线抗结核药物,已被指出与肝毒性有关。尽管数十年来进行了广泛研究,但INH诱导肝毒性的机制仍知之甚少。最近,线粒体毒性已成为INH诱导肝毒性的一种新范式。在本研究中,我们表明INH损害了人肝癌HepG2细胞中的线粒体生物发生和动力学。INH降低了线粒体膜电位(MMP)并诱导线粒体肿胀。INH还抑制了三种主要线粒体生物发生调节因子SIRT1、PGC1α和NRF1的蛋白表达,同时PGC1α的乙酰化增加。与此同时,INH减少了线粒体数量,伴随着线粒体蛋白COX IV表达的降低。INH导致线粒体碎片化,涉及融合蛋白MFN2以及裂变蛋白DRP1水平的降低。INH降低的DRP1表达与细胞凋亡增加有关,表明存在促生存裂变及其参与线粒体质量控制。INH激活了p38丝裂原活化蛋白激酶(p38 MAPK),而抑制p38 MAPK会加重INH诱导的SIRT1、PGC1α、NRF1、COX IV和DRP1表达的降低。抑制p38 MAPK还进一步上调了PGC1α的乙酰化,并加剧了INH诱导的MMP丧失、线粒体肿胀和细胞凋亡。综上所述,INH激活的p38 MAPK通过部分恢复SIRT1 - PGC1α途径激活来诱导线粒体生物发生以减轻细胞凋亡。与此同时,INH激活的p38 MAPK通过部分恢复DRP1表达促进保护性线粒体裂变以减轻细胞凋亡。