Hirsova Petra, Weng Peggy, Salim Warda, Bronk Steven F, Griffith Thomas S, Ibrahim Samar H, Gores Gregory J
Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN 55905, USA.
Department of Urology, University of Minnesota, Minneapolis, MN 55455, USA.
Hepatol Commun. 2017 Sep;1(7):648-662. doi: 10.1002/hep4.1069. Epub 2017 Jul 20.
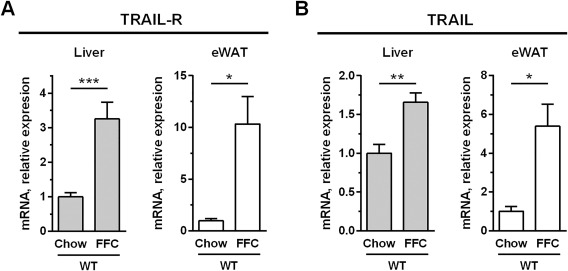
BACKGROUND & AIM: TNF-related apoptosis-inducing ligand (TRAIL) and its cognate receptor(s) are upregulated in human and murine nonalcoholic steatohepatitis (NASH). However, the consequence of this enhanced expression on NASH pathogenesis remains unclear. TRAIL may either accentuate liver injury by promoting hepatic steatosis and inflammation, or it may mitigate the disease process by improving systemic insulin resistance and averting hepatic fibrosis. Herein, we investigated the role of TRAIL in an obesity-induced murine model of NASH.
C57BL/6 wild-type (WT) mice and mice were placed on a 20-week standard chow or FFC (high fat, fructose, and cholesterol) diet which induces obesity, insulin resistance and NASH. Metabolic phenotype, liver injury, inflammation and fibrosis, and adipose tissue homeostasis were examined.
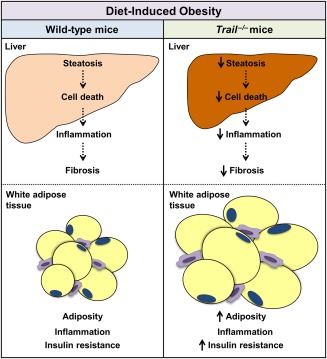
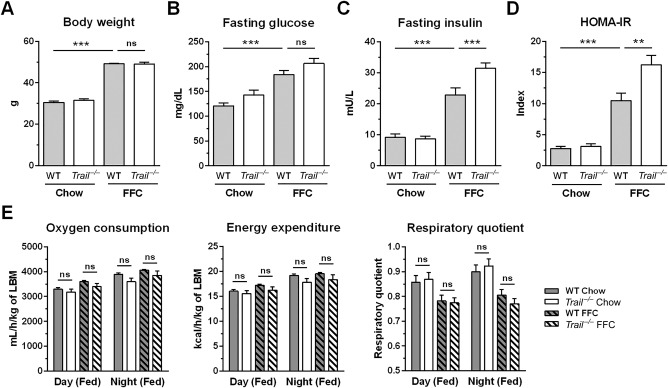
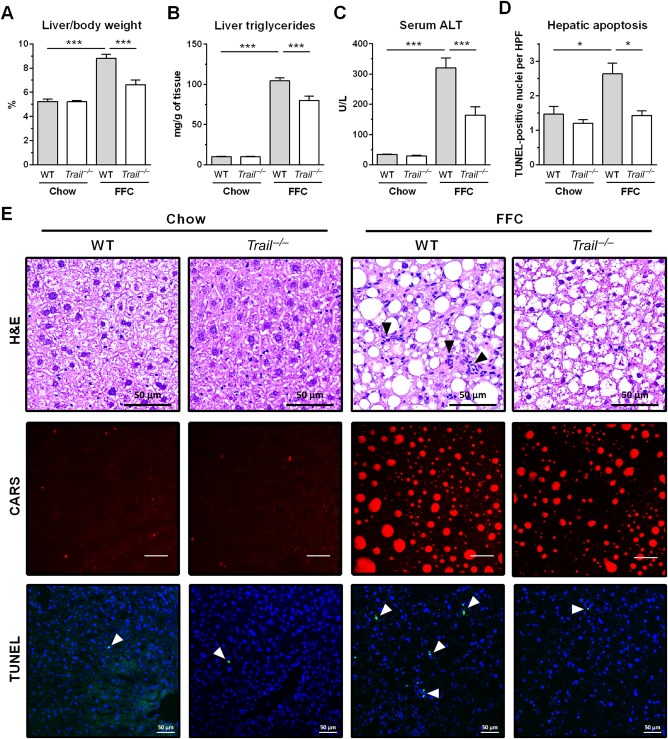
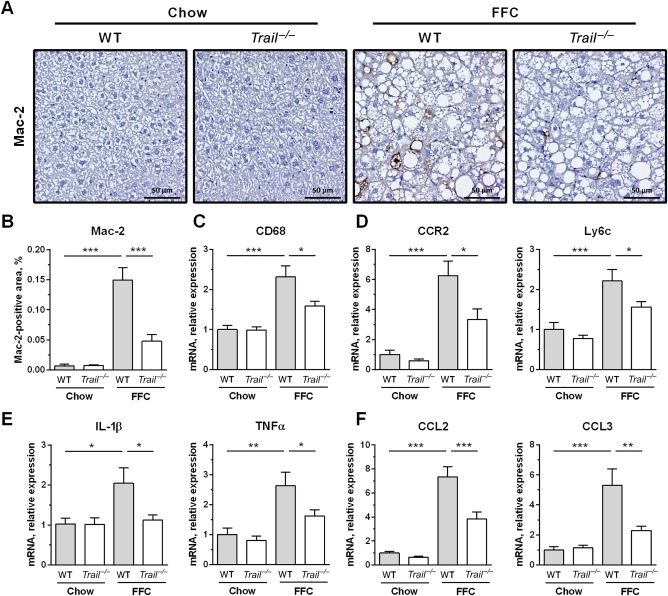
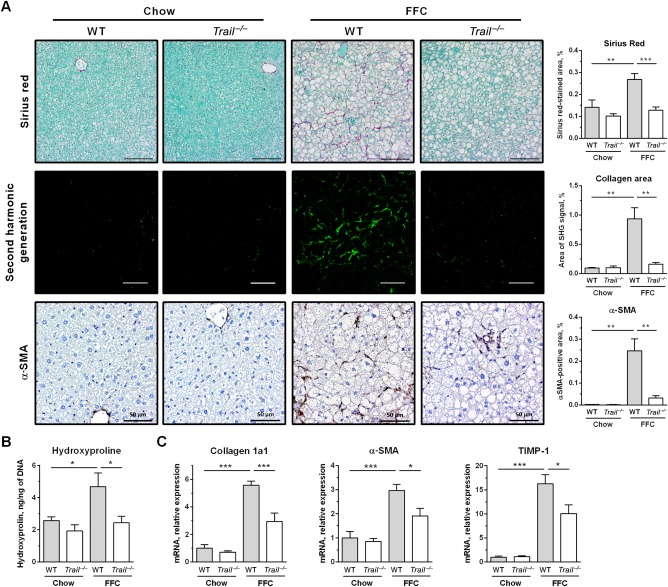
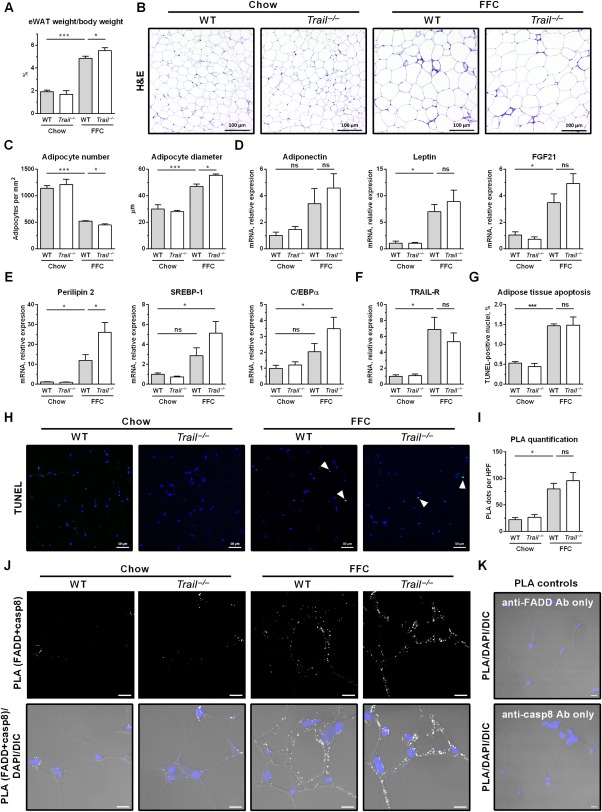
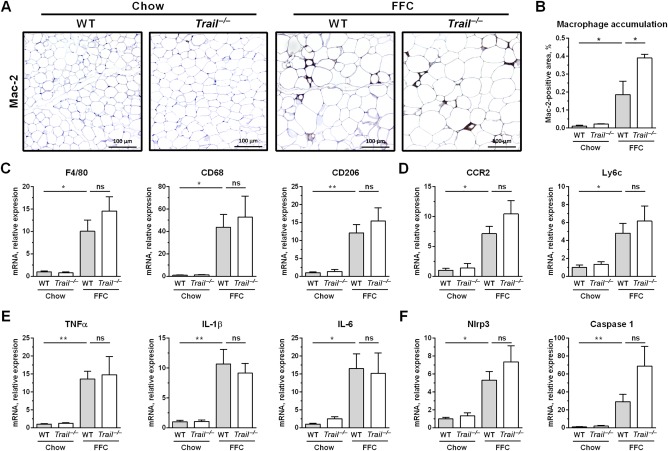
FFC diet-fed mice displayed no difference in weight gain and metabolic profile when compared to WT mice on the same diet. All FFC-fed mice developed significant hepatic steatosis, which was attenuated in mice. TRAIL deficiency also significantly decreased FFC diet-induced liver injury as manifest by reduced serum ALT values, hepatic TUNEL-positive cells and macrophage-associated inflammation. FFC diet-associated hepatic stellate cell activation and hepatic collagen deposition were also abrogated in mice. In contrast to the liver, TRAIL deletion did not improve FFC diet-induced adipose tissue injury and inflammation, and actually aggravated insulin resistance. In , these observations employing genetic TRAIL inactivation suggest that NASH pathogenesis may be dissociated from other features of the metabolic syndrome and liver-targeted inhibition of TRAIL signaling may be salutary.
肿瘤坏死因子相关凋亡诱导配体(TRAIL)及其同源受体在人类和小鼠非酒精性脂肪性肝炎(NASH)中表达上调。然而,这种表达增强对NASH发病机制的影响仍不清楚。TRAIL可能通过促进肝脏脂肪变性和炎症加重肝损伤,也可能通过改善全身胰岛素抵抗和避免肝纤维化减轻疾病进程。在此,我们研究了TRAIL在肥胖诱导的NASH小鼠模型中的作用。
将C57BL/6野生型(WT)小鼠和[此处原文缺失相关小鼠信息]小鼠置于20周的标准饲料或FFC(高脂肪、果糖和胆固醇)饮食中,该饮食可诱导肥胖、胰岛素抵抗和NASH。检测代谢表型、肝损伤、炎症和纤维化以及脂肪组织稳态。
与喂食相同饮食的WT小鼠相比,喂食FFC饮食的[此处原文缺失相关小鼠信息]小鼠在体重增加和代谢谱方面无差异。所有喂食FFC的小鼠均出现显著的肝脏脂肪变性,而在[此处原文缺失相关小鼠信息]小鼠中这种情况有所减轻。TRAIL缺乏也显著降低了FFC饮食诱导的肝损伤,表现为血清ALT值降低、肝脏TUNEL阳性细胞减少以及巨噬细胞相关炎症减轻。FFC饮食相关的肝星状细胞激活和肝脏胶原沉积在[此处原文缺失相关小鼠信息]小鼠中也被消除。与肝脏相反,TRAIL缺失并未改善FFC饮食诱导的脂肪组织损伤和炎症,实际上还加重了胰岛素抵抗。总之,这些采用基因敲除TRAIL的观察结果表明,NASH发病机制可能与代谢综合征的其他特征无关,并且对TRAIL信号的肝脏靶向抑制可能是有益的。