Mazariegos-Canellas Oriol, Do Trien, Peto Tim, Eyre David W, Underwood Anthony, Crook Derrick, Wyllie David H
Nuffield Department of Medicine, John Radcliffe Hospital, Headley Way, Oxford, OX3 9DU, UK.
Public Health England, 61 Colindale Avenue, London, NW9 5EQ, UK.
BMC Bioinformatics. 2017 Nov 13;18(1):477. doi: 10.1186/s12859-017-1907-2.
Large scale bacterial sequencing has made the determination of genetic relationships within large sequence collections of bacterial genomes derived from the same microbial species an increasingly common task. Solutions to the problem have application to public health (for example, in the detection of possible disease transmission), and as part of divide-and-conquer strategies selecting groups of similar isolates for computationally intensive methods of phylogenetic inference using (for example) maximal likelihood methods. However, the generation and maintenance of distance matrices is computationally intensive, and rapid methods of doing so are needed to allow translation of microbial genomics into public health actions.
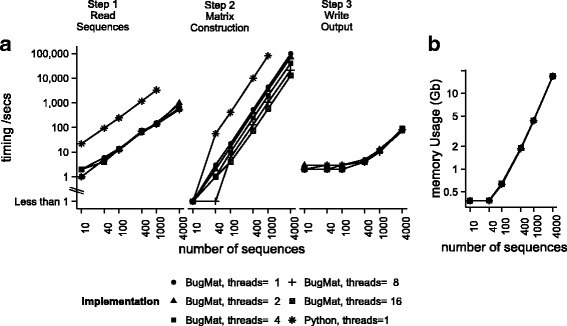
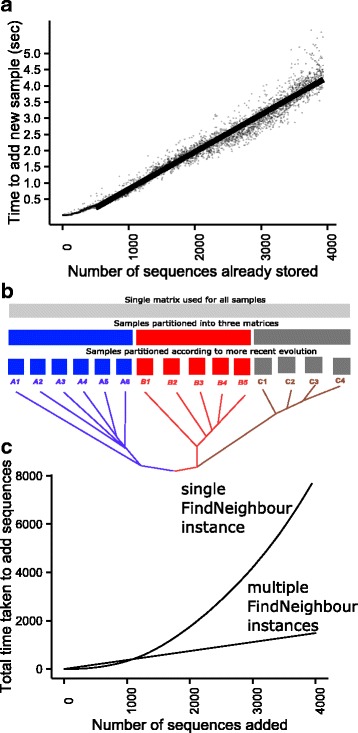
We developed, tested and deployed three solutions. BugMat is a fast C++ application which generates one-off in-memory distance matrices. FindNeighbour and FindNeighbour2 are server-side applications which build, maintain, and persist either complete (for FindNeighbour) or sparse (for FindNeighbour2) distance matrices given a set of sequences. FindNeighbour and BugMat use a variation model to accelerate computation, while FindNeighbour2 uses reference-based compression. Performance metrics show scalability into tens of thousands of sequences, with options for scaling further.
Three applications, each with distinct strengths and weaknesses, are available for distance-matrix based analysis of large bacterial collections. Deployed as part of the Public Health England solution for M. tuberculosis genomic processing, they will have wide applicability.
大规模细菌测序已使确定源自同一微生物物种的大量细菌基因组序列集合中的遗传关系成为一项日益常见的任务。该问题的解决方案可应用于公共卫生领域(例如,检测可能的疾病传播),并且作为分而治之策略的一部分,选择相似分离株群体用于使用(例如)最大似然法等计算密集型系统发育推断方法。然而,距离矩阵的生成和维护计算量很大,因此需要快速方法来实现将微生物基因组学转化为公共卫生行动。
我们开发、测试并部署了三种解决方案。BugMat是一个快速的C++应用程序,可一次性生成内存中的距离矩阵。FindNeighbour和FindNeighbour2是服务器端应用程序,给定一组序列后,它们可以构建、维护并持久化完整的(用于FindNeighbour)或稀疏的(用于FindNeighbour2)距离矩阵。FindNeighbour和BugMat使用变异模型来加速计算,而FindNeighbour2使用基于参考的压缩。性能指标表明可扩展到数万个序列,并且还有进一步扩展的选项。
有三种应用程序可用于对大量细菌集合进行基于距离矩阵的分析,每种应用程序都有各自的优缺点。作为英国公共卫生部门结核分枝杆菌基因组处理解决方案的一部分进行部署,它们将具有广泛的适用性。