Big Data Institute, University of Oxford, Oxford, United Kingdom
National Institute for Health Research Oxford Biomedical Research Centre, Oxford, United Kingdom.
J Clin Microbiol. 2019 Dec 23;58(1). doi: 10.1128/JCM.01037-19.
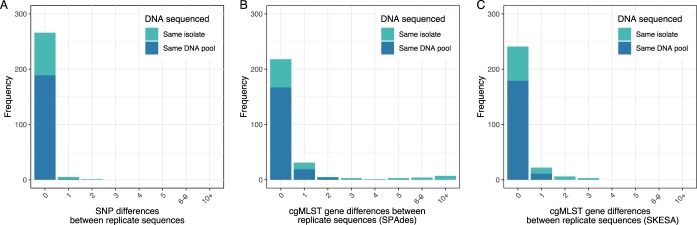
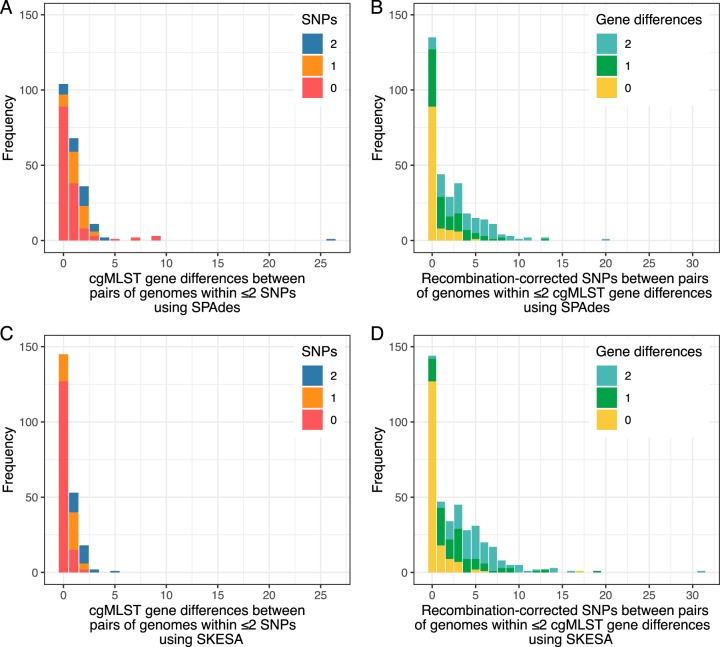
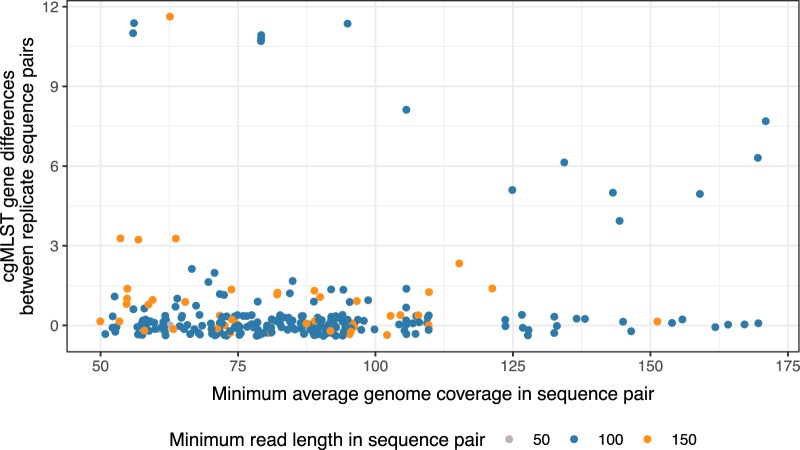
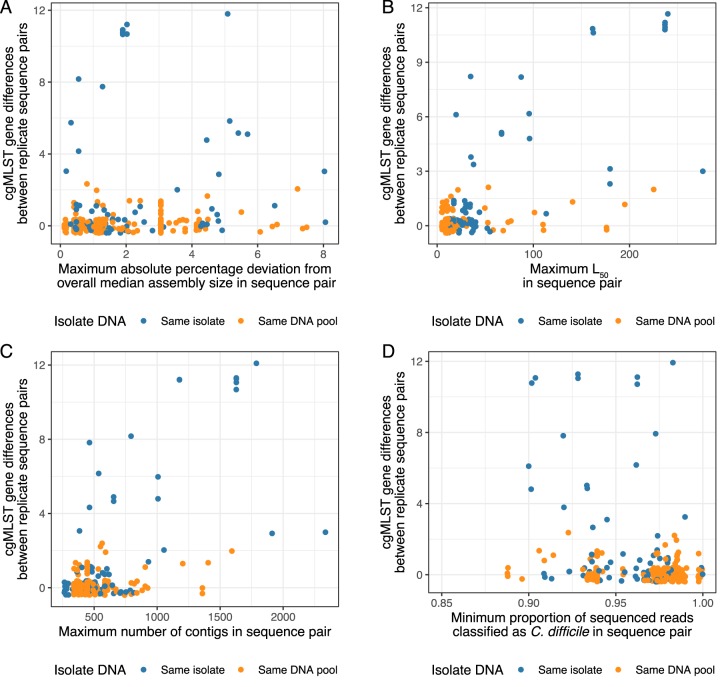
Pathogen whole-genome sequencing has huge potential as a tool to better understand infection transmission. However, rapidly identifying closely related genomes among a background of thousands of other genomes is challenging. Here, we describe a refinement to core genome multilocus sequence typing (cgMLST) in which alleles at each gene are reproducibly converted to a unique hash, or short string of letters (hash-cgMLST). This avoids the resource-intensive need for a single centralized database of sequentially numbered alleles. We test the reproducibility and discriminatory power of cgMLST/hash-cgMLST compared to those of mapping-based approaches in , using repeated sequencing of the same isolates (replicates) and data from consecutive infection isolates from six English hospitals. Hash-cgMLST provided the same results as standard cgMLST, with minimal performance penalty. Comparing 272 replicate sequence pairs using reference-based mapping, there were 0, 1, or 2 single-nucleotide polymorphisms (SNPs) between 262 (96%), 5 (2%), and 1 (<1%) of the pairs, respectively. Using hash-cgMLST, 218 (80%) of replicate pairs assembled with SPAdes had zero gene differences, and 31 (11%), 5 (2%), and 18 (7%) pairs had 1, 2, and >2 differences, respectively. False gene differences were clustered in specific genes and associated with fragmented assemblies, but were reduced using the SKESA assembler. Considering 412 pairs of infections with ≤2 SNPS, i.e., consistent with recent transmission, 376 (91%) had ≤2 gene differences and 16 (4%) had ≥4. Comparing a genome to 100,000 others took <1 min using hash-cgMLST. Hash-cgMLST is an effective surveillance tool for rapidly identifying clusters of related genomes. However, cgMLST/hash-cgMLST generate more false variants than mapping-based approaches. Follow-up mapping-based analyses are likely required to precisely define close genetic relationships.
病原体全基因组测序作为一种更好地了解感染传播的工具具有巨大的潜力。然而,在数以千计的其他基因组背景下快速识别密切相关的基因组是具有挑战性的。在这里,我们描述了一种核心基因组多位点序列分型(cgMLST)的改进方法,其中每个基因的等位基因可重复转换为唯一的哈希值或短字母串(哈希-cgMLST)。这避免了对顺序编号等位基因的单一集中式数据库的资源密集型需求。我们使用来自英国六家医院的连续感染分离株的数据,在 1000 个重复分离株和 6 个连续感染分离株中重复测序(重复)来测试 cgMLST/hash-cgMLST 与基于映射方法的重现性和区分能力。哈希-cgMLST 提供了与标准 cgMLST 相同的结果,性能损失最小。使用基于参考的映射比较 272 对重复序列对,在 262 对(96%)、5 对(2%)和 1 对(<1%)的对中分别存在 0、1 或 2 个单核苷酸多态性(SNP)。使用 hash-cgMLST,使用 SPAdes 组装的 218 对(80%)重复对具有零个基因差异,而 31 对(11%)、5 对(2%)和 18 对(7%)具有 1、2 和 >2 个差异,分别。假基因差异聚类在特定基因中,并与碎片化组装相关,但使用 SKESA 组装器可减少这些差异。考虑到 412 对 SNP 差异≤2 的感染,即与近期传播一致,376 对(91%)具有≤2 个基因差异,16 对(4%)具有≥4 个基因差异。使用 hash-cgMLST 比较一个基因组与 100000 个其他基因组不到 1 分钟。Hash-cgMLST 是一种快速识别相关基因组簇的有效监测工具。然而,与基于映射的方法相比,cgMLST/hash-cgMLST 会产生更多的假变体。可能需要后续基于映射的分析来精确定义密切的遗传关系。