Ifowonco Inc, Vancouver, Canada.
Genome Sciences Centre, BC Cancer Agency, Vancouver, Canada.
J Comput Aided Mol Des. 2018 Jan;32(1):299-311. doi: 10.1007/s10822-017-0085-7. Epub 2017 Nov 13.
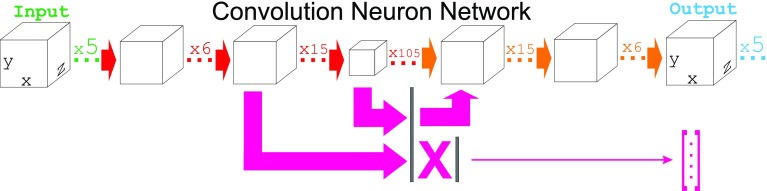
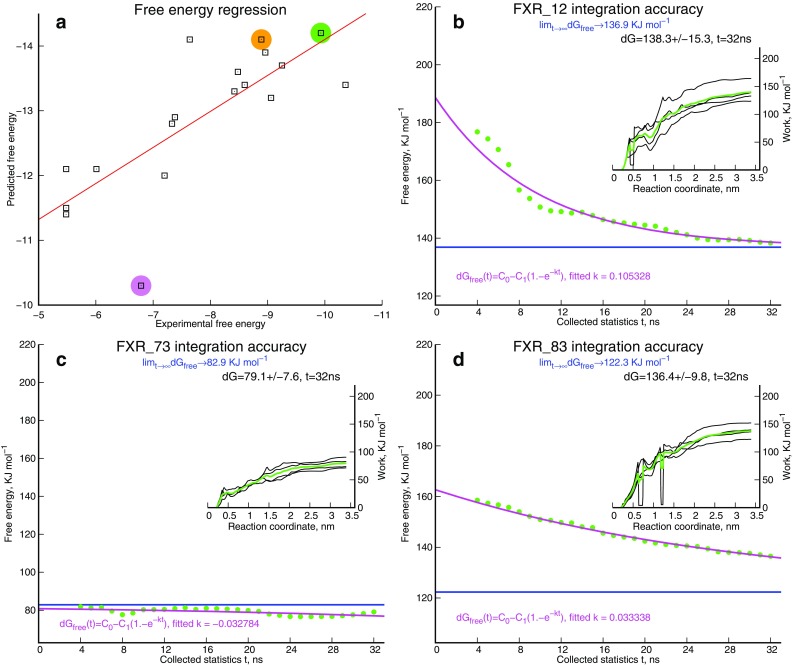
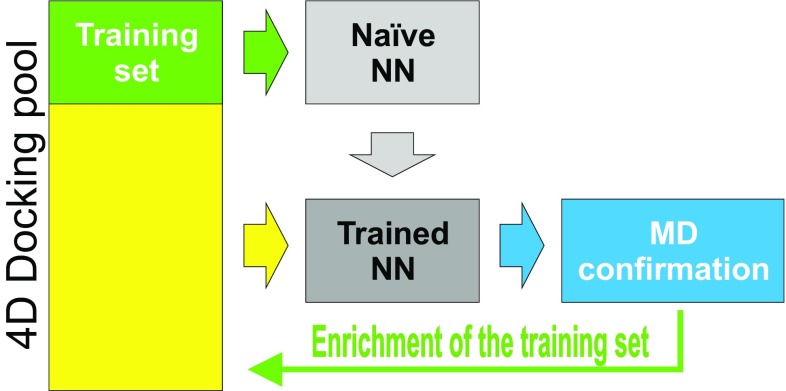
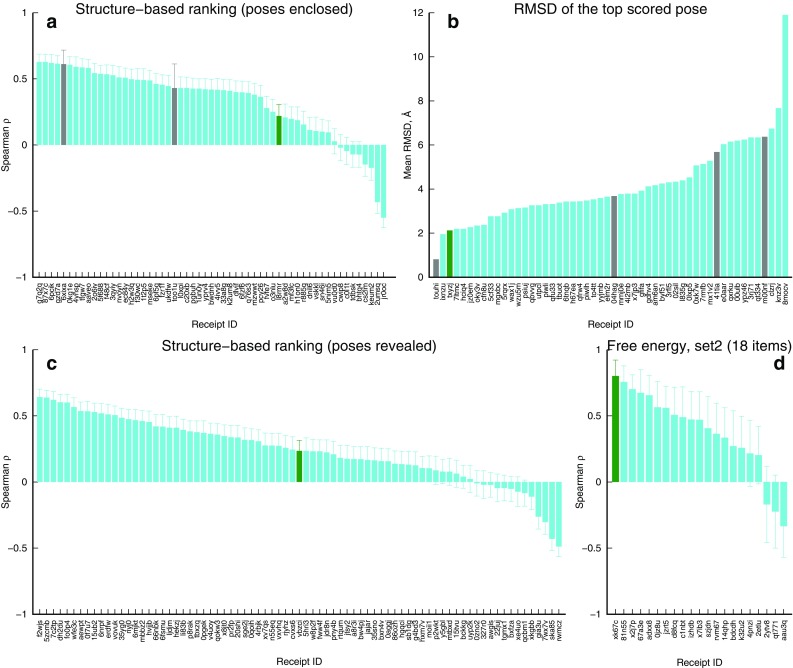
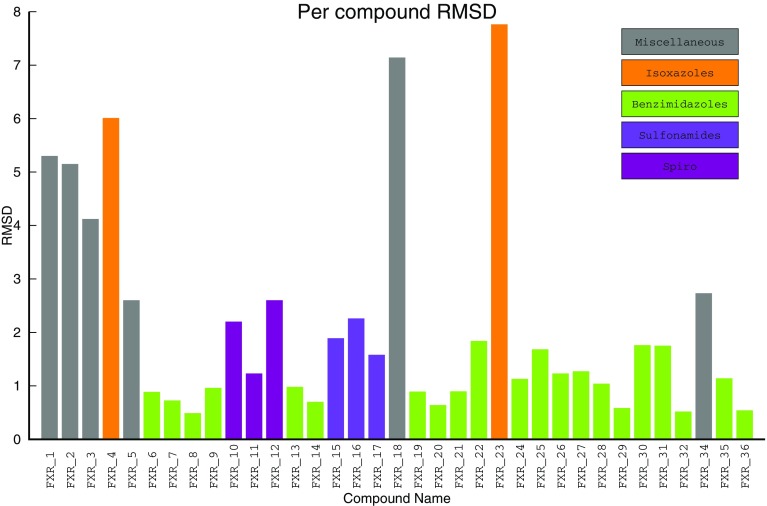
We report the implementation of molecular modeling approaches developed as a part of the 2016 Grand Challenge 2, the blinded competition of computer aided drug design technologies held by the D3R Drug Design Data Resource ( https://drugdesigndata.org/ ). The challenge was focused on the ligands of the farnesoid X receptor (FXR), a highly flexible nuclear receptor of the cholesterol derivative chenodeoxycholic acid. FXR is considered an important therapeutic target for metabolic, inflammatory, bowel and obesity related diseases (Expert Opin Drug Metab Toxicol 4:523-532, 2015), but in the context of this competition it is also interesting due to the significant ligand-induced conformational changes displayed by the protein. To deal with these conformational changes we employed multiple simulations of molecular dynamics (MD). Our MD-based protocols were top-ranked in estimating the free energy of binding of the ligands and FXR protein. Our approach was ranked second in the prediction of the binding poses where we also combined MD with molecular docking and artificial neural networks. Our approach showed mediocre results for high-throughput scoring of interactions.
我们报告了作为 2016 年大挑战 2 的一部分开发的分子建模方法的实施情况,该大挑战是由 D3R 药物设计数据资源(https://drugdesigndata.org/)举办的计算机辅助药物设计技术的盲赛。该挑战的重点是法尼醇 X 受体(FXR)的配体,法尼醇 X 受体是胆固醇衍生物鹅去氧胆酸的高度灵活的核受体。FXR 被认为是代谢、炎症、肠道和肥胖相关疾病的重要治疗靶点(Expert Opin Drug Metab Toxicol 4:523-532, 2015),但在本次竞争中,由于该蛋白显示出显著的配体诱导构象变化,因此也很有趣。为了处理这些构象变化,我们采用了多次分子动力学(MD)模拟。我们基于 MD 的方案在估算配体和 FXR 蛋白的结合自由能方面排名最高。我们的方法在预测结合构象方面排名第二,我们还将 MD 与分子对接和人工神经网络相结合。我们的方法在高吞吐量评分方面表现中等。