Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children's Hospital, Westmead, New South Wales, Australia.
Sydney Medical School, University of Sydney, Sydney, New South Wales, Australia.
J Neurol Neurosurg Psychiatry. 2018 Feb;89(2):127-137. doi: 10.1136/jnnp-2017-316880. Epub 2017 Nov 15.
We characterised the clinical course, treatment and outcomes in 59 patients with relapsing myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination.
We evaluated clinical phenotypes, annualised relapse rates (ARR) prior and on immunotherapy and Expanded Disability Status Scale (EDSS), in 218 demyelinating episodes from 33 paediatric and 26 adult patients.
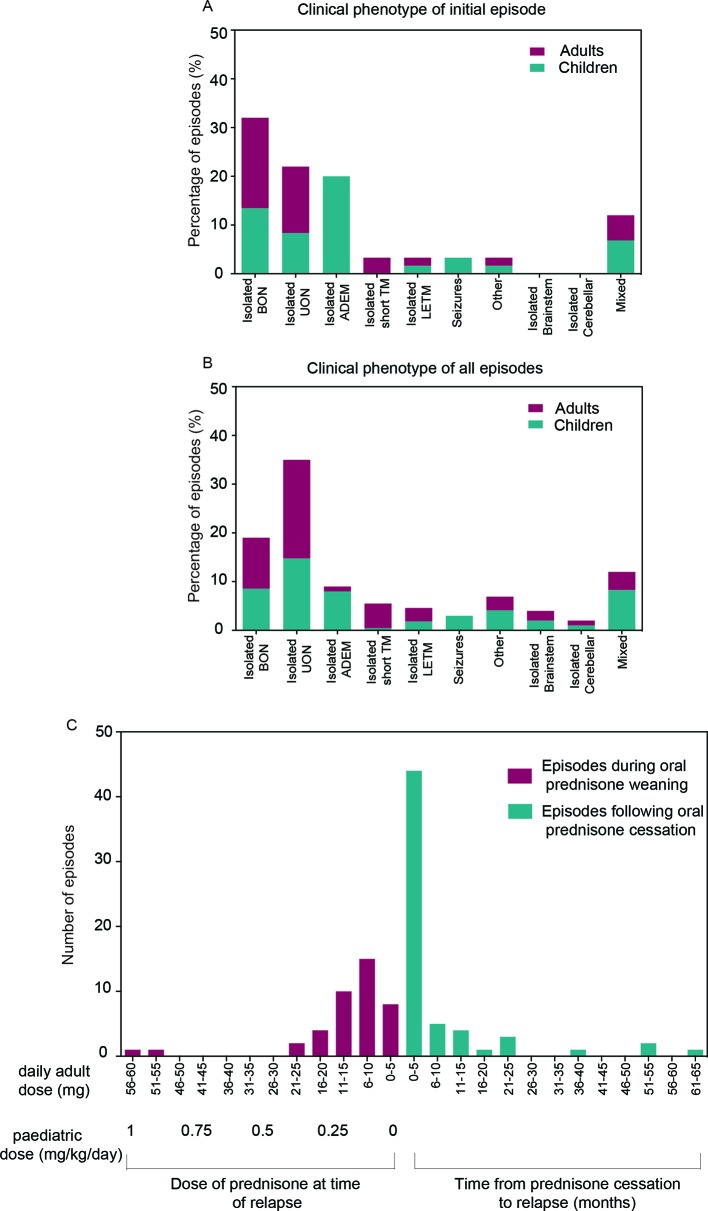
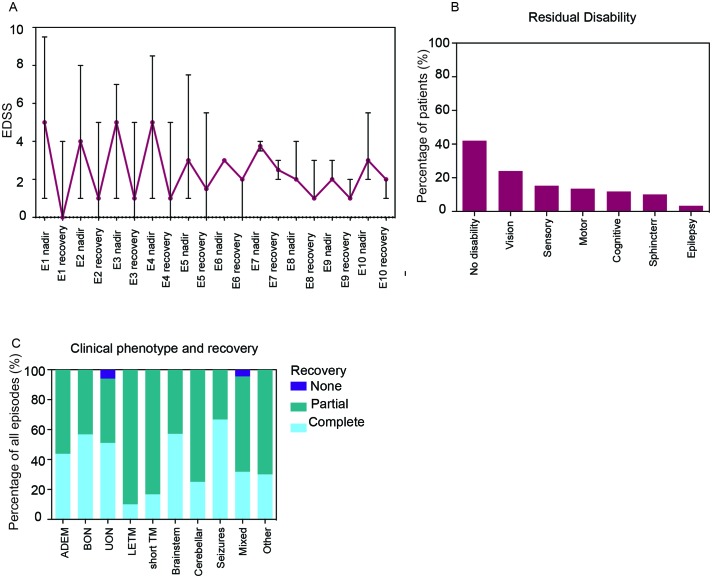
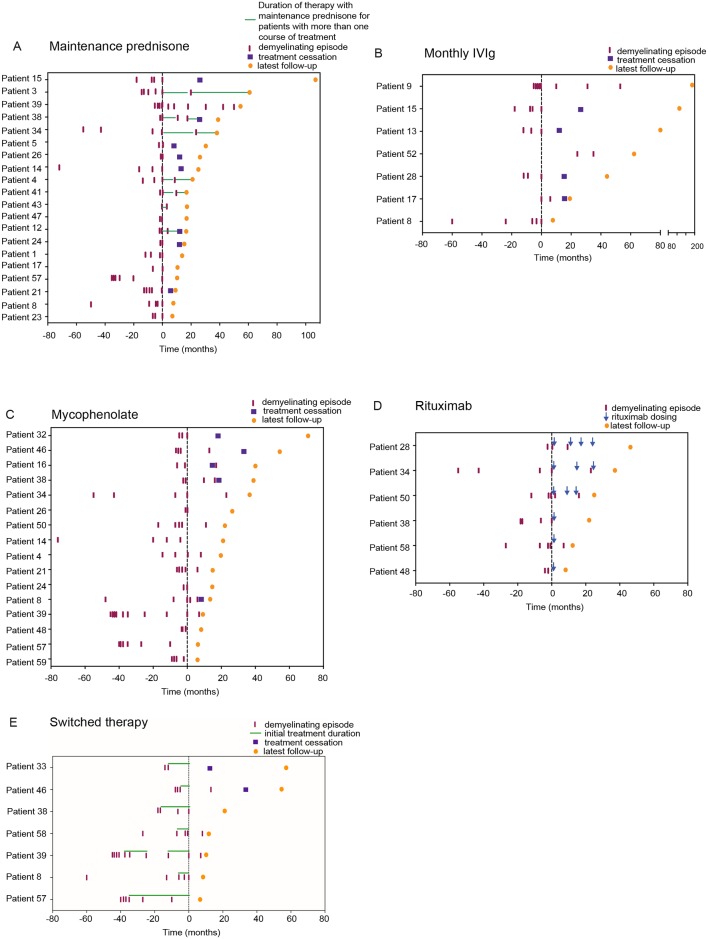
The most common initial presentation in the cohort was optic neuritis (ON) in 54% (bilateral (BON) 32%, unilateral (UON) 22%), followed by acute disseminated encephalomyelitis (ADEM) (20%), which occurred exclusively in children. ON was the dominant phenotype (UON 35%, BON 19%) of all clinical episodes. 109/226 (48%) MRIs had no brain lesions. Patients were steroid responsive, but 70% of episodes treated with oral prednisone relapsed, particularly at doses <10 mg daily or within 2 months of cessation. Immunotherapy, including maintenance prednisone (P=0.0004), intravenous immunoglobulin, rituximab and mycophenolate, all reduced median ARRs on-treatment. Treatment failure rates were lower in patients on maintenance steroids (5%) compared with non-steroidal maintenance immunotherapy (38%) (P=0.016). 58% of patients experienced residual disability (average follow-up 61 months, visual loss in 24%). Patients with ON were less likely to have sustained disability defined by a final EDSS of ≥2 (OR 0.15, P=0.032), while those who had any myelitis were more likely to have sustained residual deficits (OR 3.56, P=0.077).
Relapsing MOG antibody-associated demyelination is strongly associated with ON across all age groups and ADEM in children. Patients are highly responsive to steroids, but vulnerable to relapse on steroid reduction and cessation.
我们描述了 59 例复发型髓鞘少突胶质细胞糖蛋白(MOG)抗体相关性脱髓鞘患者的临床病程、治疗方法和结局。
我们评估了 33 名儿科和 26 名成年患者的 218 次脱髓鞘发作中的临床表型、免疫治疗前和治疗后的年复发率(ARR)以及扩展残疾状况量表(EDSS)评分。
该队列中最常见的初始表现为视神经炎(ON),占 54%(双侧(BON)32%,单侧(UON)22%),其次是急性播散性脑脊髓炎(ADEM),占 20%,仅发生于儿童。ON 是所有临床发作的主要表型(UON 35%,BON 19%)。226 次 MRI 中有 109 次无脑部病变。患者对类固醇治疗有反应,但 70%接受口服泼尼松治疗的发作会复发,尤其是在剂量<10mg/d 或停药后 2 个月内。免疫治疗,包括维持性泼尼松(P=0.0004)、静脉注射免疫球蛋白、利妥昔单抗和霉酚酸酯,均可降低治疗期间的中位 ARR。维持性类固醇治疗(5%)的治疗失败率低于非类固醇维持性免疫治疗(38%)(P=0.016)。58%的患者有持续性残疾(平均随访 61 个月,24%有视力丧失)。ON 患者发生持续性残疾(定义为最终 EDSS≥2)的可能性较小(OR 0.15,P=0.032),而任何脊髓炎患者发生持续性残余缺陷的可能性较大(OR 3.56,P=0.077)。
复发型 MOG 抗体相关性脱髓鞘与所有年龄组的 ON 以及儿童的 ADEM 密切相关。患者对类固醇高度敏感,但在类固醇减量和停药时易复发。