Andersson Emma R, Chivukula Indira V, Hankeova Simona, Sjöqvist Marika, Tsoi Yat Long, Ramsköld Daniel, Masek Jan, Elmansuri Aiman, Hoogendoorn Anita, Vazquez Elenae, Storvall Helena, Netušilová Julie, Huch Meritxell, Fischler Björn, Ellis Ewa, Contreras Adriana, Nemeth Antal, Chien Kenneth C, Clevers Hans, Sandberg Rickard, Bryja Vitezslav, Lendahl Urban
Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden; Department of Biosciences and Nutrition, Karolinska Institutet, Stockholm, Sweden.
Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden; Current affiliation: Integrated Cardio Metabolic Centre (ICMC), Karolinska Institutet, Huddinge, Sweden.
Gastroenterology. 2018 Mar;154(4):1080-1095. doi: 10.1053/j.gastro.2017.11.002. Epub 2017 Nov 21.
BACKGROUND & AIMS: Alagille syndrome is a genetic disorder characterized by cholestasis, ocular abnormalities, characteristic facial features, heart defects, and vertebral malformations. Most cases are associated with mutations in JAGGED1 (JAG1), which encodes a Notch ligand, although it is not clear how these contribute to disease development. We aimed to develop a mouse model of Alagille syndrome to elucidate these mechanisms.
Mice with a missense mutation (H268Q) in Jag1 (Jag1 mice) were outbred to a C3H/C57bl6 background to generate a mouse model for Alagille syndrome (Jag1 mice). Liver tissues were collected at different timepoints during development, analyzed by histology, and liver organoids were cultured and analyzed. We performed transcriptome analysis of Jag1 livers and livers from patients with Alagille syndrome, cross-referenced to the Human Protein Atlas, to identify commonly dysregulated pathways and biliary markers. We used species-specific transcriptome separation and ligand-receptor interaction assays to measure Notch signaling and the ability of JAG1 to bind or activate Notch receptors. We studied signaling of JAG1 and JAG1 via NOTCH 1, NOTCH2, and NOTCH3 and resulting gene expression patterns in parental and NOTCH1-expressing C2C12 cell lines.
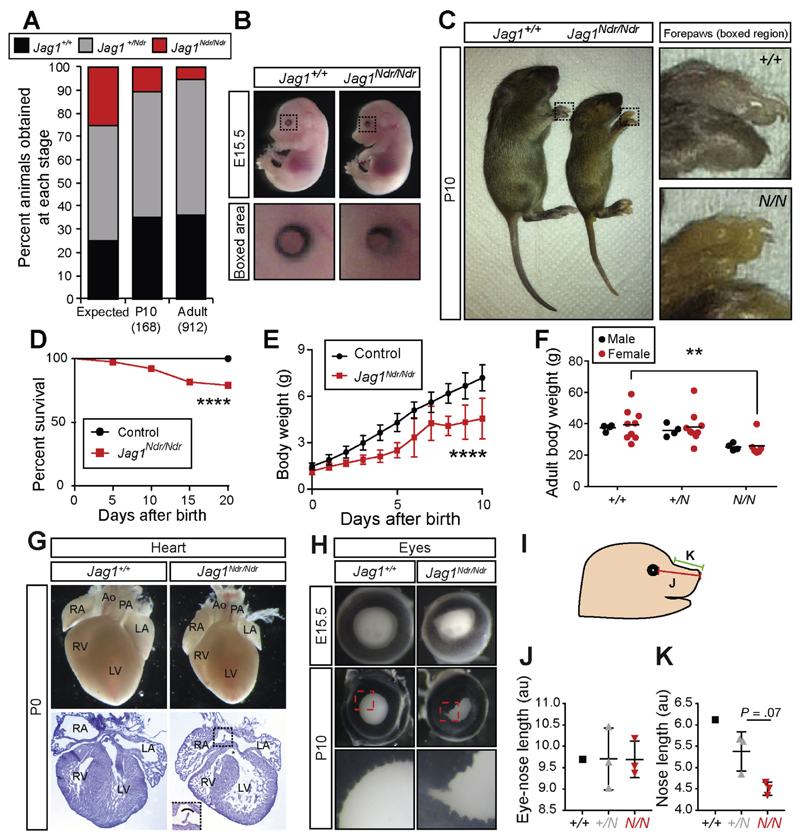
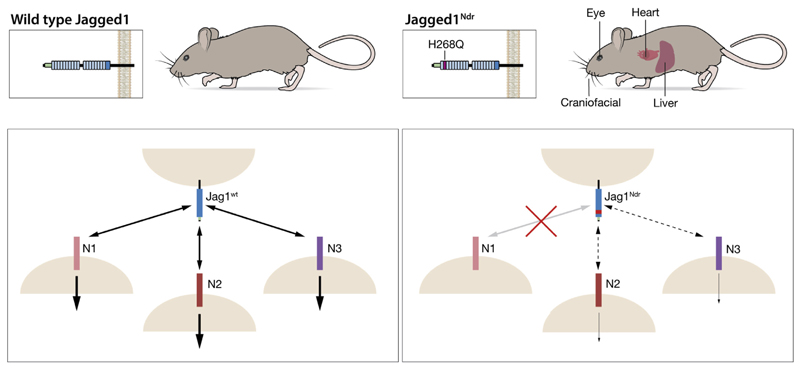
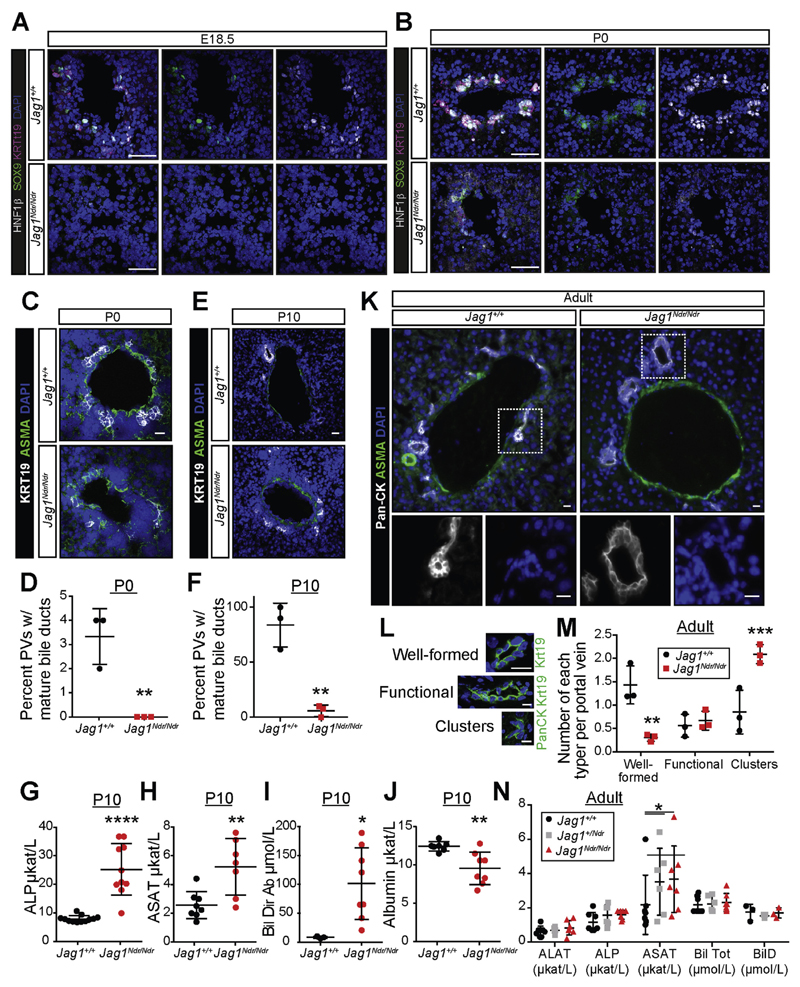
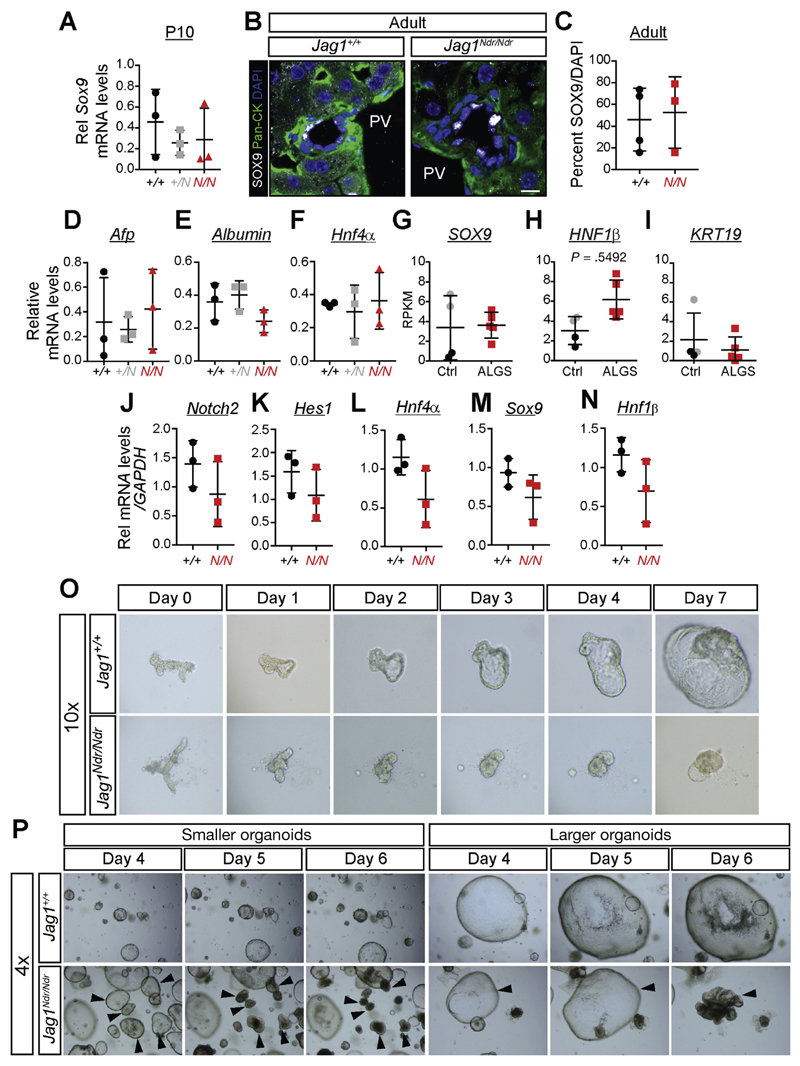
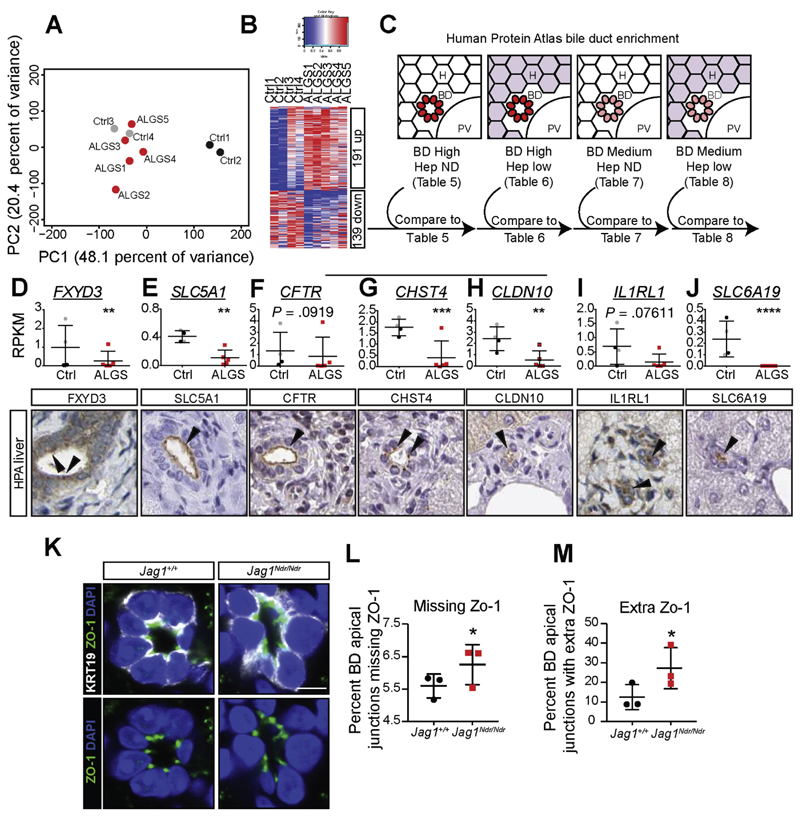
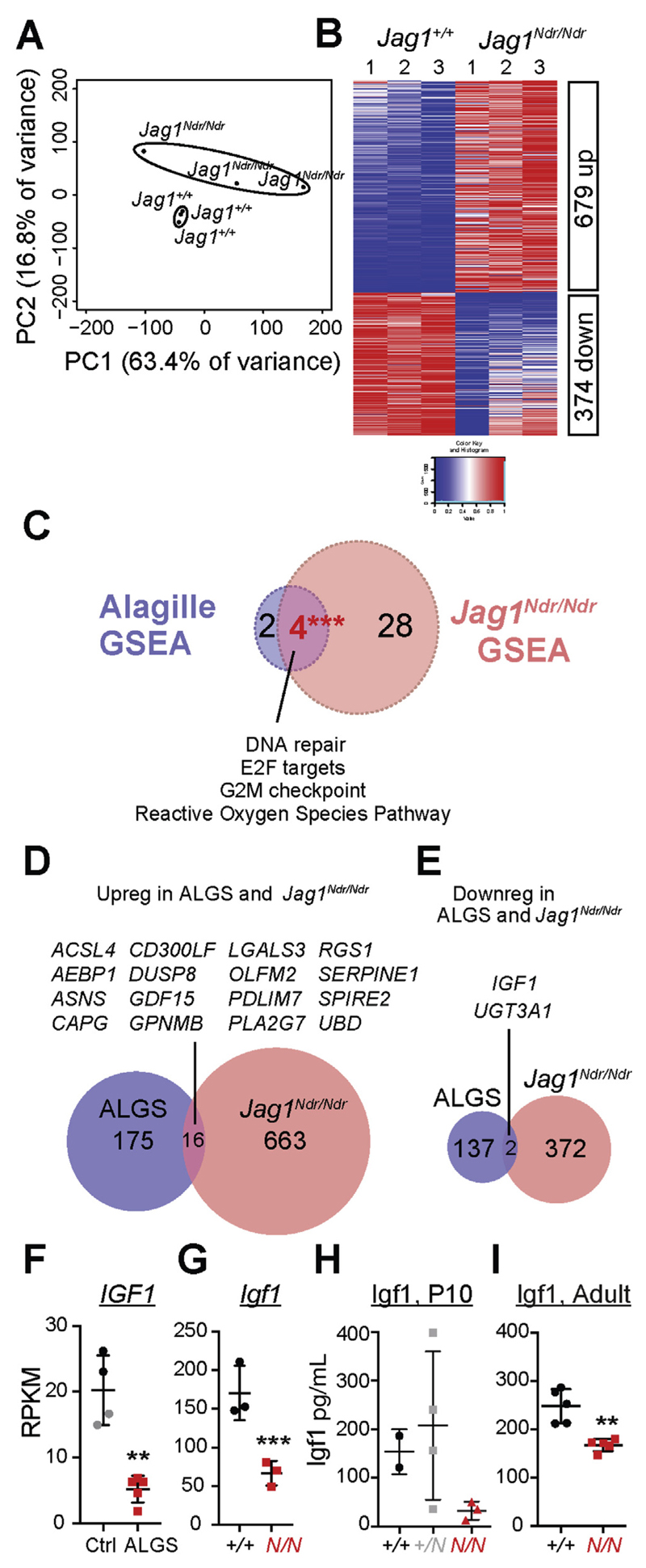
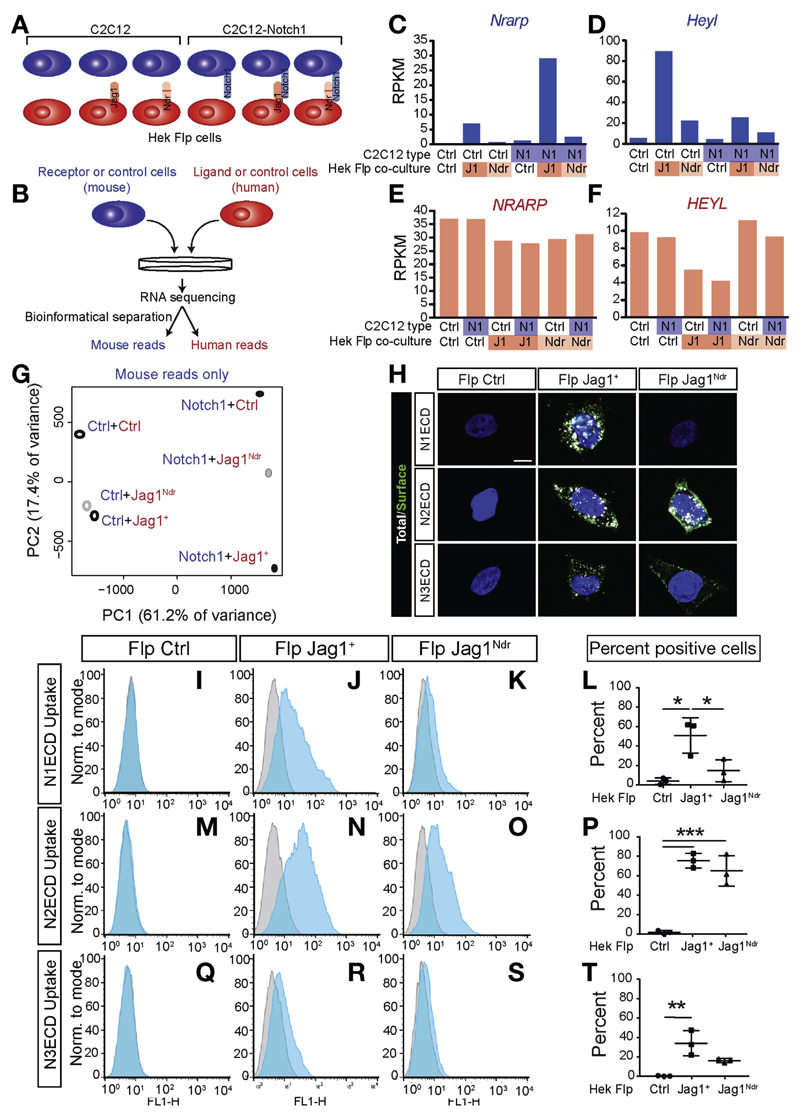
Jag1 mice had many features of Alagille syndrome, including eye, heart, and liver defects. Bile duct differentiation, morphogenesis, and function were dysregulated in newborn Jag1 mice, with aberrations in cholangiocyte polarity, but these defects improved in adult mice. Jag1 liver organoids collapsed in culture, indicating structural instability. Whole-transcriptome sequence analyses of liver tissues from mice and patients with Alagille syndrome identified dysregulated genes encoding proteins enriched at the apical side of cholangiocytes, including CFTR and SLC5A1, as well as reduced expression of IGF1. Exposure of Notch-expressing cells to JAG1, compared with JAG1, led to hypomorphic Notch signaling, based on transcriptome analysis. JAG1-expressing cells, but not JAG1-expressing cells, bound soluble Notch1 extracellular domain, quantified by flow cytometry. However, JAG1 and JAG1 cells each bound NOTCH2, and signaling from NOTCH2 signaling was reduced but not completely inhibited, in response to JAG1 compared with JAG1.
In mice, expression of a missense mutant of Jag1 (Jag1) disrupts bile duct development and recapitulates Alagille syndrome phenotypes in heart, eye, and craniofacial dysmorphology. JAG1 does not bind NOTCH1, but binds NOTCH2, and elicits hypomorphic signaling. This mouse model can be used to study other features of Alagille syndrome and organ development.
阿拉吉耶综合征是一种遗传性疾病,其特征为胆汁淤积、眼部异常、特征性面部特征、心脏缺陷和脊柱畸形。大多数病例与编码Notch配体的JAGGED1(JAG1)基因突变有关,尽管尚不清楚这些突变如何导致疾病发展。我们旨在建立一种阿拉吉耶综合征小鼠模型以阐明这些机制。
将Jag1基因存在错义突变(H268Q)的小鼠(Jag1小鼠)与C3H/C57bl6背景的小鼠杂交,以生成阿拉吉耶综合征小鼠模型(Jag1小鼠)。在发育的不同时间点收集肝脏组织,进行组织学分析,并培养和分析肝脏类器官。我们对Jag1小鼠肝脏和阿拉吉耶综合征患者的肝脏进行转录组分析,并与人类蛋白质图谱交叉参考,以确定共同失调的通路和胆管标志物。我们使用物种特异性转录组分离和配体-受体相互作用测定来测量Notch信号传导以及JAG1结合或激活Notch受体的能力。我们研究了JAG1和JAG1通过NOTCH 1、NOTCH2和NOTCH3的信号传导以及在亲本和表达NOTCH1的C2C12细胞系中产生的基因表达模式。
Jag1小鼠具有阿拉吉耶综合征的许多特征,包括眼睛、心脏和肝脏缺陷。新生Jag1小鼠的胆管分化、形态发生和功能失调,胆管细胞极性异常,但这些缺陷在成年小鼠中有所改善。Jag1肝脏类器官在培养中塌陷,表明结构不稳定。对小鼠和阿拉吉耶综合征患者肝脏组织的全转录组序列分析确定,编码在胆管细胞顶端侧富集的蛋白质的基因失调,包括CFTR和SLC5A1,以及IGF1表达降低。基于转录组分析,与JAG1相比,将表达Notch的细胞暴露于JAG1会导致Notch信号传导减弱。通过流式细胞术定量分析,表达JAG1的细胞而非表达JAG1的细胞结合可溶性Notch1细胞外结构域。然而,JAG1和JAG1细胞均结合NOTCH2,与JAG1相比,响应JAG1时NOTCH2信号传导减弱但未完全抑制。
在小鼠中,Jag1(Jag1)错义突变体的表达破坏胆管发育,并在心脏、眼睛和颅面畸形方面重现阿拉吉耶综合征表型。JAG1不结合NOTCH1,但结合NOTCH2,并引发减弱的信号传导。这种小鼠模型可用于研究阿拉吉耶综合征的其他特征和器官发育。