Soraggi Samuele, Wiuf Carsten, Albrechtsen Anders
Department of Mathematical Sciences, Faculty of Science, University of Copenhagen, 2100, Denmark
Department of Mathematical Sciences, Faculty of Science, University of Copenhagen, 2100, Denmark.
G3 (Bethesda). 2018 Feb 2;8(2):551-566. doi: 10.1534/g3.117.300192.
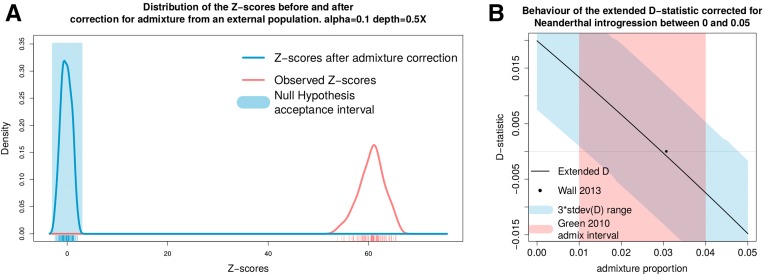
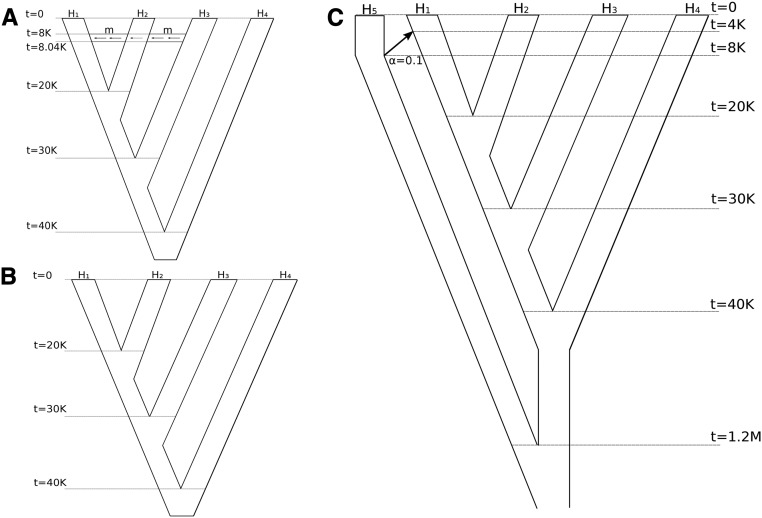
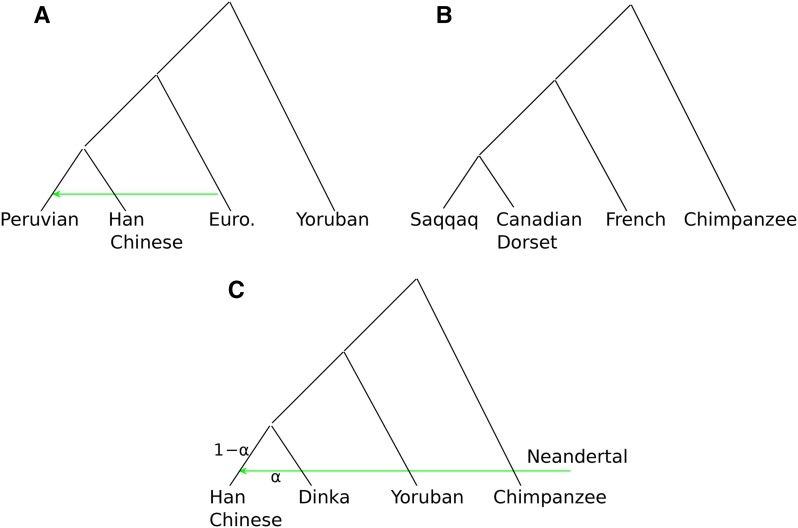
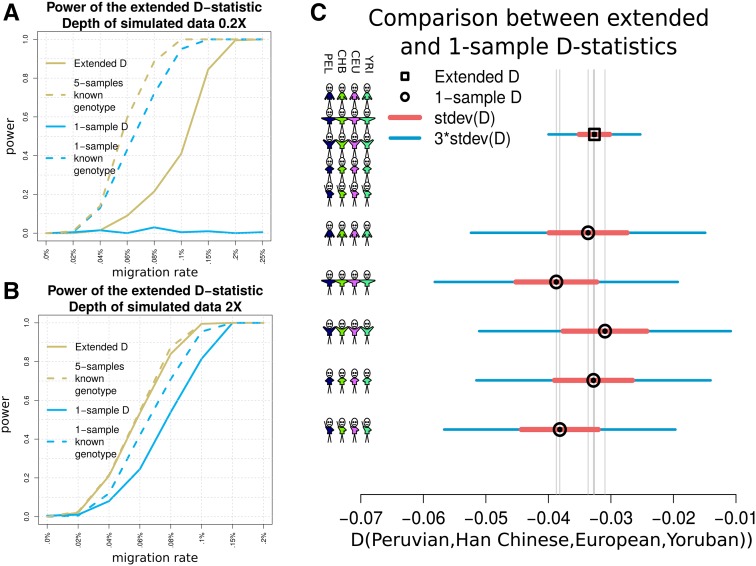
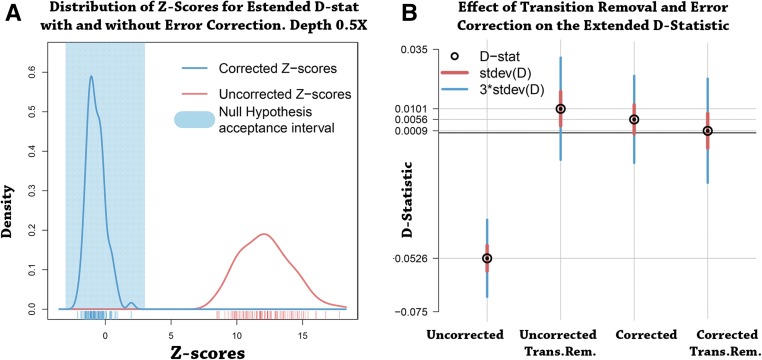
The detection of ancient gene flow between human populations is an important issue in population genetics. A common tool for detecting ancient admixture events is the D-statistic. The D-statistic is based on the hypothesis of a genetic relationship that involves four populations, whose correctness is assessed by evaluating specific coincidences of alleles between the groups. When working with high-throughput sequencing data, calling genotypes accurately is not always possible; therefore, the D-statistic currently samples a single base from the reads of one individual per population. This implies ignoring much of the information in the data, an issue especially striking in the case of ancient genomes. We provide a significant improvement to overcome the problems of the D-statistic by considering all reads from multiple individuals in each population. We also apply type-specific error correction to combat the problems of sequencing errors, and show a way to correct for introgression from an external population that is not part of the supposed genetic relationship, and how this leads to an estimate of the admixture rate. We prove that the D-statistic is approximated by a standard normal distribution. Furthermore, we show that our method outperforms the traditional D-statistic in detecting admixtures. The power gain is most pronounced for low and medium sequencing depth (1-10×), and performances are as good as with perfectly called genotypes at a sequencing depth of 2×. We show the reliability of error correction in scenarios with simulated errors and ancient data, and correct for introgression in known scenarios to estimate the admixture rates.
检测人类群体间的古代基因流动是群体遗传学中的一个重要问题。检测古代混合事件的常用工具是D统计量。D统计量基于一种涉及四个群体的遗传关系假设,其正确性通过评估群体间等位基因的特定巧合来评估。在处理高通量测序数据时,准确地调用基因型并不总是可行的;因此,D统计量目前从每个群体中一个个体的读数中采样单个碱基。这意味着忽略了数据中的许多信息,在古代基因组的情况下,这个问题尤为突出。我们通过考虑每个群体中多个个体的所有读数,对克服D统计量的问题有了显著改进。我们还应用类型特异性错误校正来应对测序错误问题,并展示了一种校正来自不属于假定遗传关系的外部群体的基因渗入的方法,以及这如何导致混合率的估计。我们证明D统计量近似于标准正态分布。此外,我们表明我们的方法在检测混合方面优于传统的D统计量。在低和中等测序深度(1 - 10×)时,功率增益最为明显,在2×的测序深度下,性能与完美调用基因型时一样好。我们在具有模拟错误和古代数据的场景中展示了错误校正的可靠性,并在已知场景中校正基因渗入以估计混合率。