Thomas Milton, Fenske Gavin John, Antony Linto, Ghimire Sudeep, Welsh Ronald, Ramachandran Akhilesh, Scaria Joy
Department of Veterinary and Biomedical Sciences, South Dakota State University, Brookings, SD 57007 USA.
South Dakota Center for Biologics Research and Commercialization, Brookings, SD 57007 USA.
Gut Pathog. 2017 Nov 21;9:66. doi: 10.1186/s13099-017-0213-x. eCollection 2017.
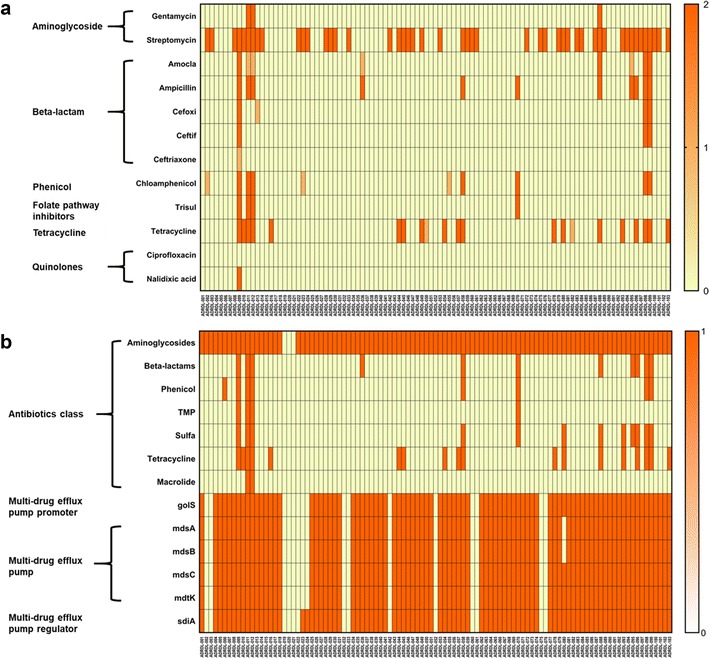
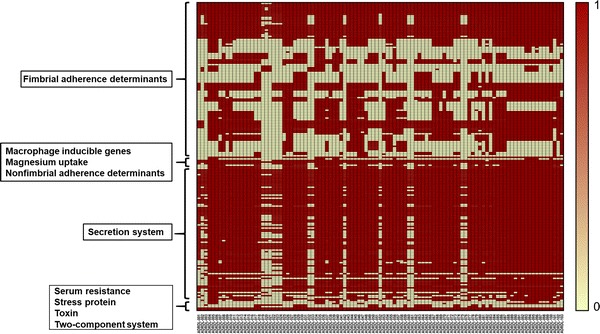
The aim of this study was to generate a reference set of genomes isolated from wildlife from the United States and to determine the antimicrobial resistance and virulence gene profile of the isolates from the genome sequence data. We sequenced the whole genomes of 103 isolates sampled between 1988 and 2003 from wildlife and exotic pet cases that were submitted to the Oklahoma Animal Disease Diagnostic Laboratory, Stillwater, Oklahoma. Among 103 isolates, 50.48% were from wild birds, 0.9% was from fish, 24.27% each were from reptiles and mammals. 50.48% isolates showed resistance to at least one antibiotic. Resistance against the aminoglycoside streptomycin was most common while 9 isolates were found to be multi-drug resistant having resistance against more than three antibiotics. Determination of virulence gene profile revealed that the genes belonging to operons, the genes that encode for type 1 fimbriae and the genes belonging to type III secretion system were predominant among the isolates. The universal presence of fimbrial genes and the genes encoded by pathogenicity islands 1-2 among the isolates we report here indicates that these isolates could potentially cause disease in humans. Therefore, the genomes we report here could be a valuable reference point for future traceback investigations when wildlife is considered to be the potential source of human Salmonellosis.
本研究的目的是生成一组从美国野生动物中分离出的基因组参考集,并根据基因组序列数据确定分离株的抗菌药物耐药性和毒力基因谱。我们对1988年至2003年间从野生动物和外来宠物病例中采集的103株分离株的全基因组进行了测序,这些病例被提交至俄克拉荷马州斯蒂尔沃特市的俄克拉荷马动物疾病诊断实验室。在103株分离株中,50.48%来自野生鸟类,0.9%来自鱼类,24.27%分别来自爬行动物和哺乳动物。50.48%的分离株对至少一种抗生素耐药。对氨基糖苷类链霉素的耐药最为常见,同时发现9株分离株具有多重耐药性,对三种以上抗生素耐药。毒力基因谱的测定显示,属于操纵子的基因、编码1型菌毛的基因以及属于III型分泌系统的基因在分离株中占主导地位。我们在此报告的分离株中普遍存在菌毛基因和致病岛1-2编码的基因,这表明这些分离株可能会导致人类疾病。因此,我们在此报告的基因组可能是未来当野生动物被认为是人类沙门氏菌病潜在来源时进行溯源调查的宝贵参考点。