Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA, USA.
Department of Biological Sciences, Vanderbilt University, Nashville, TN, USA.
Mol Ecol. 2018 Apr;27(8):1874-1883. doi: 10.1111/mec.14460. Epub 2017 Dec 29.
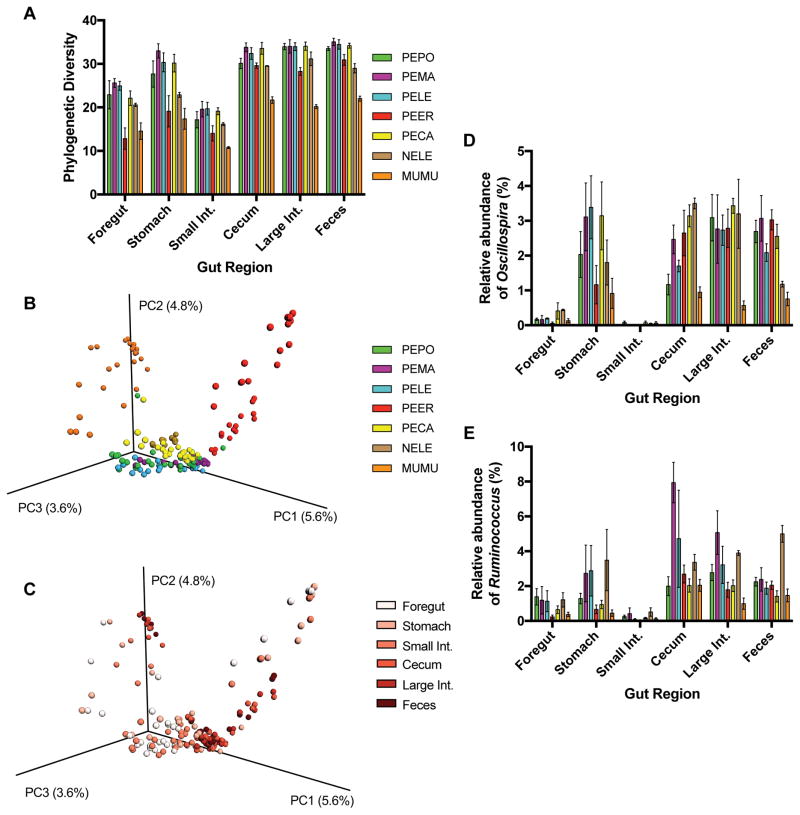
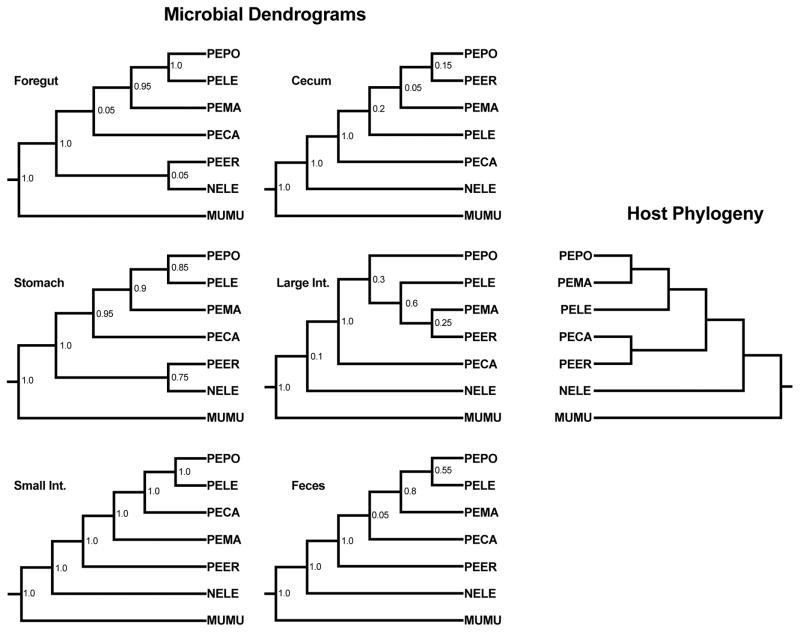
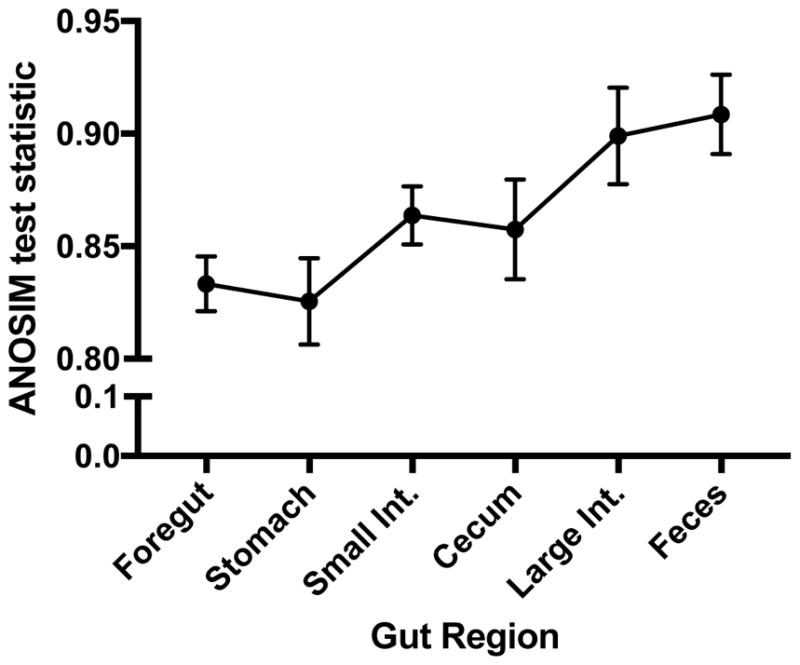
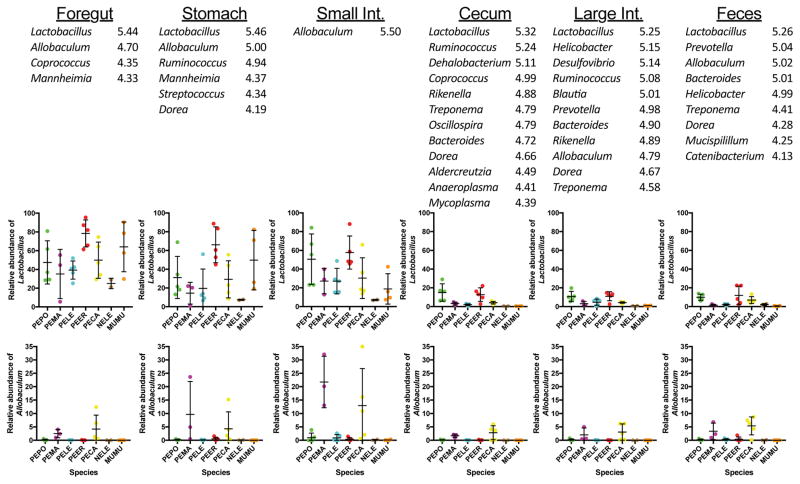
Host-associated microbial communities consist of stable and transient members that can assemble through purely stochastic processes associated with the environment or by interactions with the host. Phylosymbiosis predicts that if host-microbiota interactions impact assembly patterns, then one conceivable outcome is concordance between host evolutionary histories (phylogeny) and the ecological similarities in microbial community structures (microbiota dendrogram). This assembly pattern has been demonstrated in several clades of animal hosts in laboratory and natural populations, but in vertebrates, it has only been investigated using samples from faeces or the distal colon. Here, we collected the contents of five gut regions from seven rodent species and inventoried the bacterial communities by sequencing the 16S rRNA gene. We investigated how community structures varied across gut regions and whether the pattern of phylosymbiosis was present along the length of the gut. Gut communities varied by host species and gut region, with Oscillospira and Ruminococcus being more abundant in the stomach and hindgut regions. Gut microbial communities were highly distinguishable by host species across all gut regions, with the strength of the discrimination increasing along the length of the gut. Last, the pattern of phylosymbiosis was found in all five gut regions, as well as faeces. Aspects of the gut environment, such as oxygen levels, production of antimicrobials or other factors, may shift microbial communities across gut regions. However, regardless of these differences, host species maintain distinguishable, phylosymbiotic assemblages of microbes that may have functional impacts for the host.
宿主相关的微生物群落由稳定和瞬态成员组成,它们可以通过与环境相关的纯粹随机过程或与宿主的相互作用来组装。 共生进化预测,如果宿主-微生物群相互作用影响组装模式,那么一个可以想象的结果是宿主进化历史(系统发育)与微生物群落结构(微生物树状图)的生态相似性之间的一致性。 这种组装模式已经在动物宿主的几个分支中在实验室和自然群体中得到证明,但在脊椎动物中,仅使用粪便或远端结肠的样本进行了研究。 在这里,我们从七个啮齿动物物种中收集了五个肠道区域的内容,并通过测序 16S rRNA 基因来对细菌群落进行了编目。 我们研究了群落结构如何在肠道区域之间变化,以及共生进化的模式是否存在于肠道的长度上。 肠道群落因宿主物种和肠道区域而异,而梭菌属和瘤胃球菌属在胃和后肠区域更为丰富。 肠道微生物群落可以根据宿主物种在所有肠道区域中高度区分,随着肠道长度的增加,区分的强度也在增加。 最后,在所有五个肠道区域以及粪便中都发现了共生进化的模式。 肠道环境的各个方面,例如氧气水平、抗生素的产生或其他因素,可能会使微生物群落在肠道区域之间转移。 但是,无论存在这些差异,宿主物种都保持着可区分的、共生进化的微生物组合,这可能对宿主产生功能影响。