Kešnerová Lucie, Mars Ruben A T, Ellegaard Kirsten M, Troilo Michaël, Sauer Uwe, Engel Philipp
Department of Fundamental Microbiology, University of Lausanne, Lausanne, Switzerland.
Institute of Molecular Systems Biology, ETH Zürich, Zürich, Switzerland.
PLoS Biol. 2017 Dec 12;15(12):e2003467. doi: 10.1371/journal.pbio.2003467. eCollection 2017 Dec.
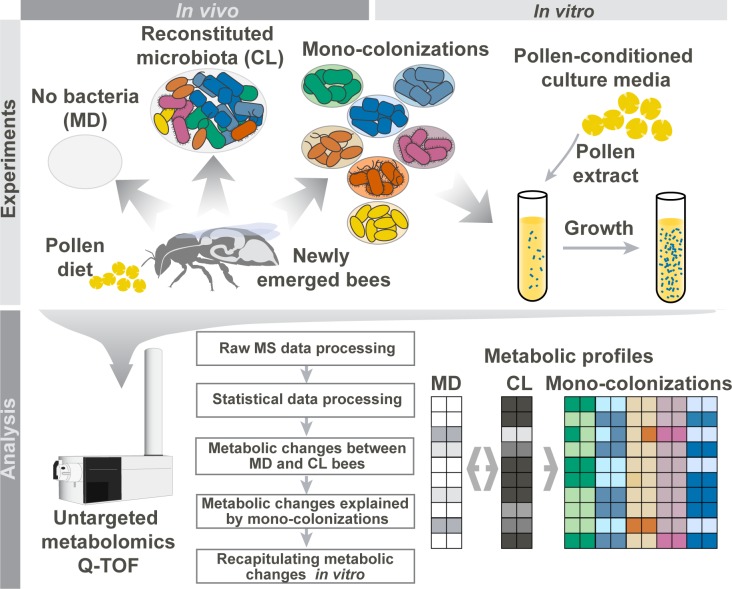
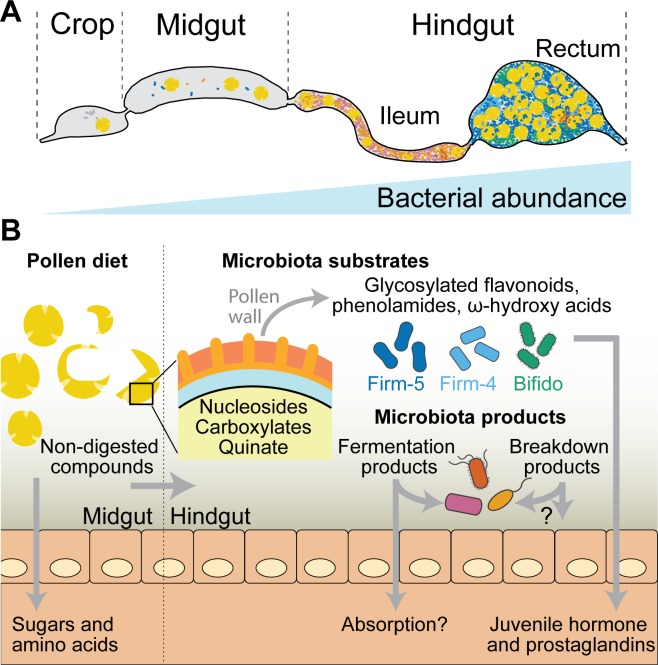
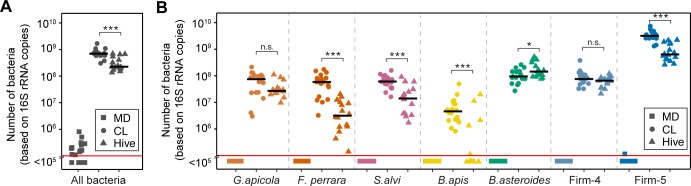
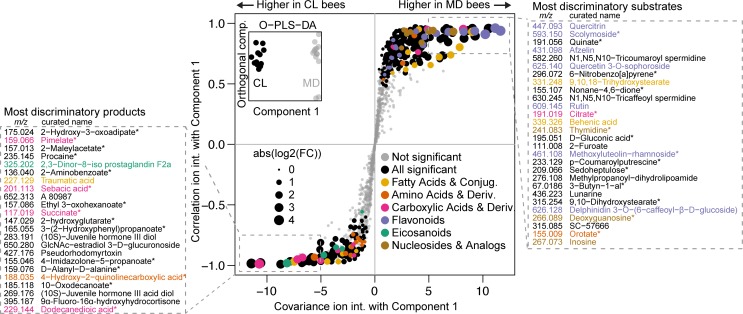
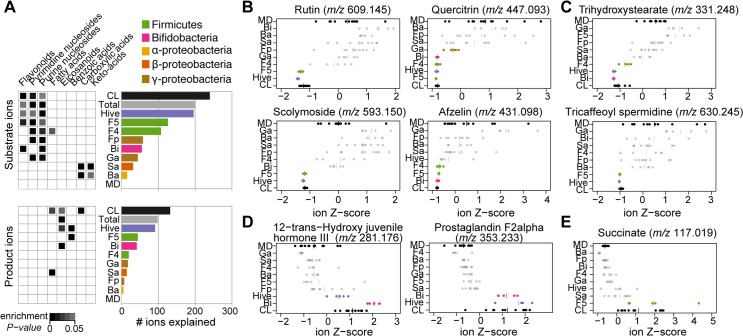
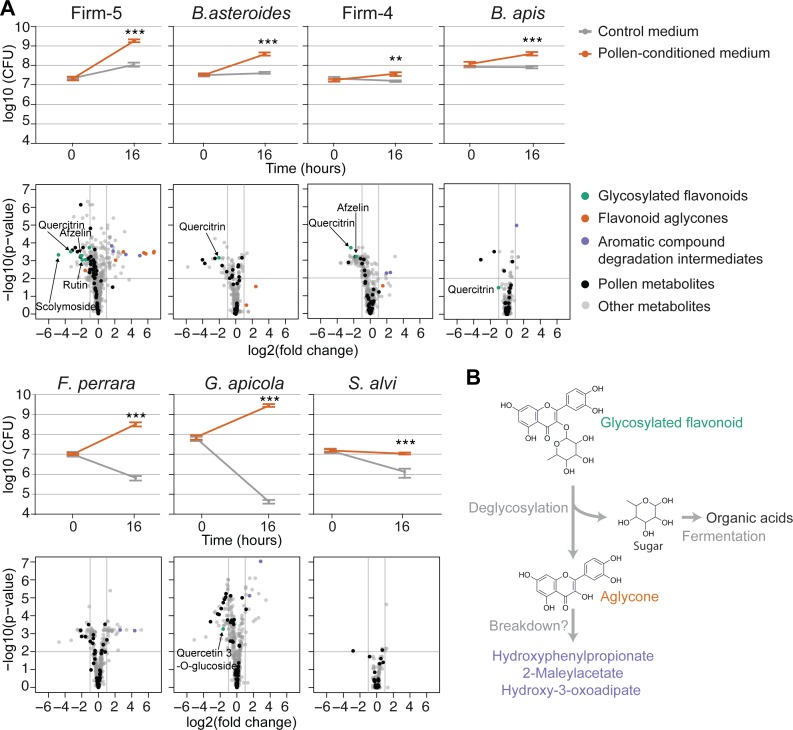
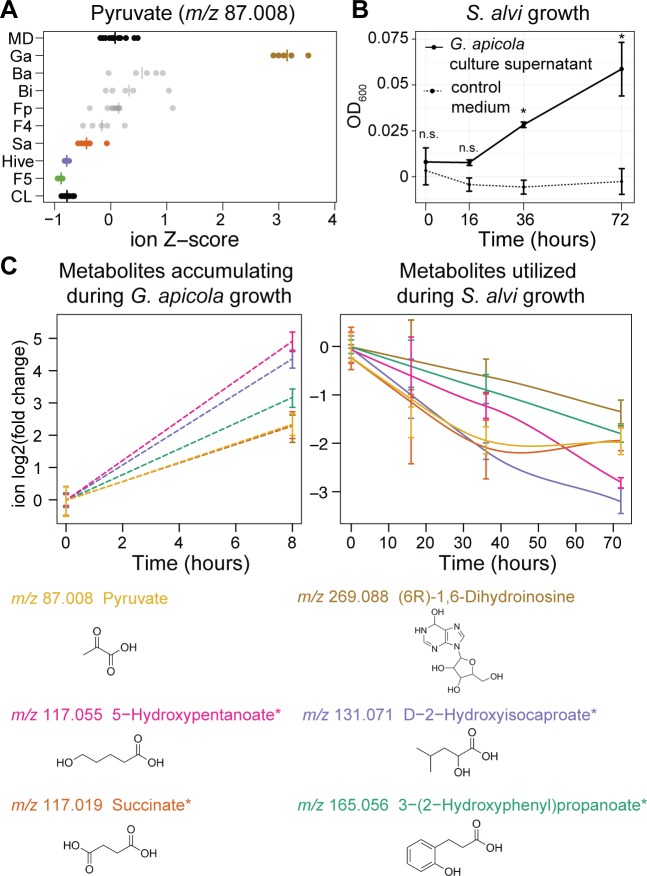
It is presently unclear how much individual community members contribute to the overall metabolic output of a gut microbiota. To address this question, we used the honey bee, which harbors a relatively simple and remarkably conserved gut microbiota with striking parallels to the mammalian system and importance for bee health. Using untargeted metabolomics, we profiled metabolic changes in gnotobiotic bees that were colonized with the complete microbiota reconstituted from cultured strains. We then determined the contribution of individual community members in mono-colonized bees and recapitulated our findings using in vitro cultures. Our results show that the honey bee gut microbiota utilizes a wide range of pollen-derived substrates, including flavonoids and outer pollen wall components, suggesting a key role for degradation of recalcitrant secondary plant metabolites and pollen digestion. In turn, multiple species were responsible for the accumulation of organic acids and aromatic compound degradation intermediates. Moreover, a specific gut symbiont, Bifidobacterium asteroides, stimulated the production of host hormones known to impact bee development. While we found evidence for cross-feeding interactions, approximately 80% of the identified metabolic changes were also observed in mono-colonized bees, with Lactobacilli being responsible for the largest share of the metabolic output. These results show that, despite prolonged evolutionary associations, honey bee gut bacteria can independently establish and metabolize a wide range of compounds in the gut. Our study reveals diverse bacterial functions that are likely to contribute to bee health and provide fundamental insights into how metabolic activities are partitioned within gut communities.
目前尚不清楚个体群落成员对肠道微生物群的整体代谢输出有多大贡献。为了解决这个问题,我们使用了蜜蜂,其拥有相对简单且高度保守的肠道微生物群,与哺乳动物系统有显著相似之处,且对蜜蜂健康至关重要。利用非靶向代谢组学,我们分析了定殖有由培养菌株重构的完整微生物群的无菌蜜蜂的代谢变化。然后我们确定了单一定殖蜜蜂中各个群落成员的贡献,并使用体外培养重现了我们的发现。我们的结果表明,蜜蜂肠道微生物群利用多种花粉衍生底物,包括黄酮类化合物和花粉外壁成分,这表明其在难降解次生植物代谢物降解和花粉消化中起关键作用。反过来,多个物种负责有机酸和芳香化合物降解中间体的积累。此外,一种特定的肠道共生菌,小行星双歧杆菌,刺激了已知影响蜜蜂发育的宿主激素的产生。虽然我们发现了交叉喂养相互作用的证据,但在单一定殖的蜜蜂中也观察到了约80%已确定的代谢变化,其中乳酸杆菌对代谢输出的贡献最大。这些结果表明,尽管有长期的进化关联,蜜蜂肠道细菌仍能在肠道中独立建立并代谢多种化合物。我们的研究揭示了可能有助于蜜蜂健康的多种细菌功能,并为肠道群落内代谢活动如何分配提供了基本见解。