Pathogen Genomics, The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK.
Clinical Microbiology and Public Health Laboratory, National Infection Service, Cambridge University Hospitals NHS Foundation Trust, Hills Road, Cambridge, UK.
BMC Genomics. 2017 Dec 28;18(1):993. doi: 10.1186/s12864-017-4399-6.
Although Mycoplasma genitalium is a common sexually transmitted pathogen causing clinically distinct diseases both in male and females, few genomes have been sequenced up to now, due mainly to its fastidious nature and slow growth. Hence, we lack a robust phylogenetic framework to provide insights into the population structure of the species. Currently our understanding of the nature and diversity of M. genitalium relies on molecular tests targeting specific genes or regions of the genome and knowledge is limited by a general under-testing internationally. This is set against a background of drug resistance whereby M. genitalium has developed resistance to mainly all therapeutic antimicrobials.
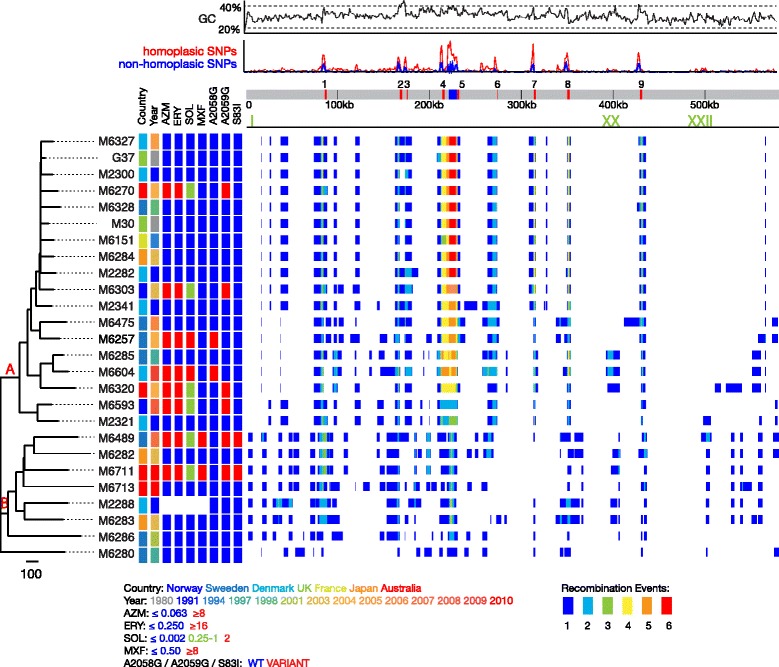
We sequenced 28 genomes of Mycoplasma genitalium from temporally (1980-2010) and geographically (Europe, Japan, Australia) diverse sources. All the strain showed essentially the same genomic content without any accessory regions found. However, we identified extensive recombination across their genomes with a total of 25 regions showing heightened levels of SNP density. These regions include the MgPar loci, associated with host interactions, as well as other genes that could also be involved in this role. Using these data, we generated a robust phylogeny which shows that there are two main clades with differing degrees of genomic variability. SNPs found in region V of 23S rRNA and parC were consistent with azithromycin/erythromycin and fluoroquinolone resistances, respectively, and with their phenotypic MIC data.
The sequence data here generated is essential for designing rational approaches to type and track Mycoplasma genitalium as antibiotic resistance increases. It represents a first approach to its population genetics to better appreciate the role of this organism as a sexually transmitted pathogen.
虽然生殖支原体是一种常见的性传播病原体,在男性和女性中都会引起明显不同的疾病,但由于其苛刻的性质和缓慢的生长速度,到目前为止只有少数基因组被测序。因此,我们缺乏一个强大的系统发育框架来深入了解该物种的种群结构。目前,我们对生殖支原体的性质和多样性的了解主要依赖于针对基因组特定基因或区域的分子检测,而国际上普遍缺乏检测,这限制了我们的认识。这种情况是在生殖支原体对主要所有治疗性抗生素产生耐药性的背景下发生的。
我们从时间(1980-2010 年)和地理(欧洲、日本、澳大利亚)上多样化的来源中对 28 个生殖支原体基因组进行了测序。所有菌株的基因组内容基本相同,没有发现任何附加区域。然而,我们在其基因组中发现了广泛的重组,共有 25 个区域显示出 SNP 密度升高。这些区域包括与宿主相互作用相关的 MgPar 基因座,以及其他可能也参与这一作用的基因。利用这些数据,我们生成了一个稳健的系统发育树,表明存在两个主要的进化枝,具有不同程度的基因组变异性。在 23S rRNA 和 parC 的区域 V 中发现的 SNP 与阿奇霉素/红霉素和氟喹诺酮类药物的耐药性分别一致,与它们的表型 MIC 数据一致。
这里生成的序列数据对于设计合理的方法来检测和追踪生殖支原体至关重要,因为抗生素耐药性不断增加。这代表了对其种群遗传学的首次探索,以更好地了解该生物体作为性传播病原体的作用。