Lee Gunhee, Lee Minho
Department of Biological Science, Sangji University, Wonju 26339, Korea.
Department of Biomedicine & Health Sciences, Graduate School, The Catholic University of Korea, Seoul 06591, Korea.
Genomics Inform. 2017 Dec;15(4):156-161. doi: 10.5808/GI.2017.15.4.156. Epub 2017 Dec 29.
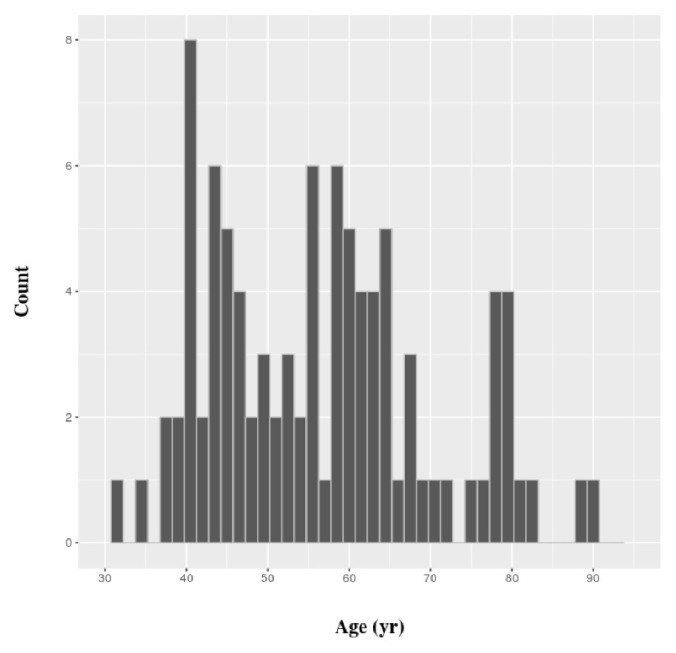
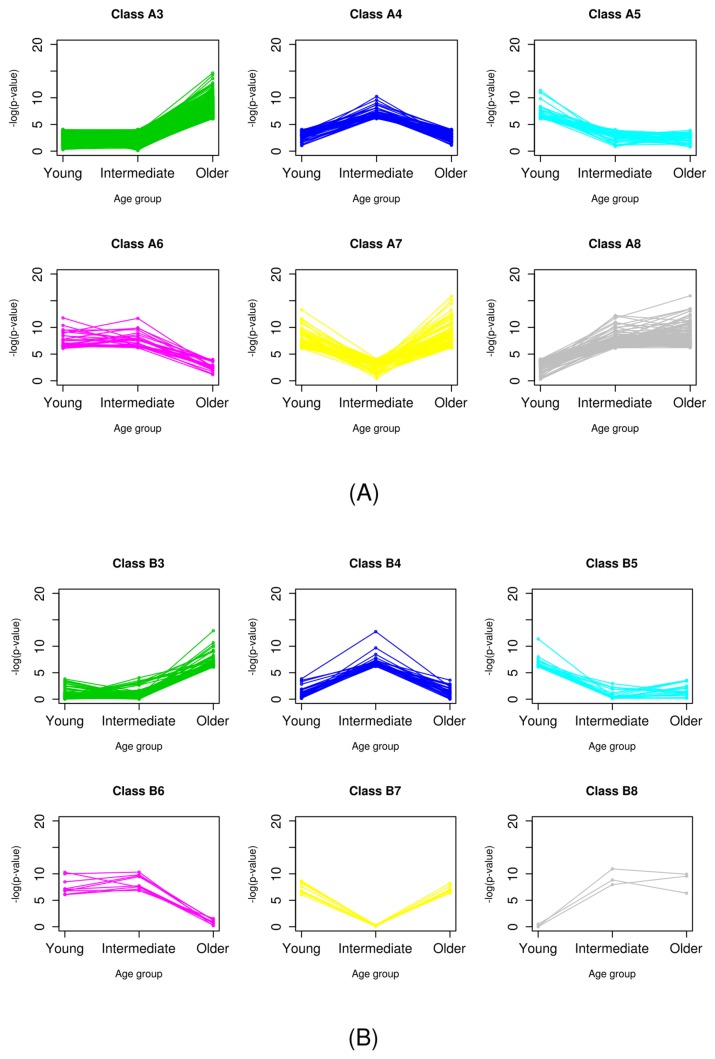
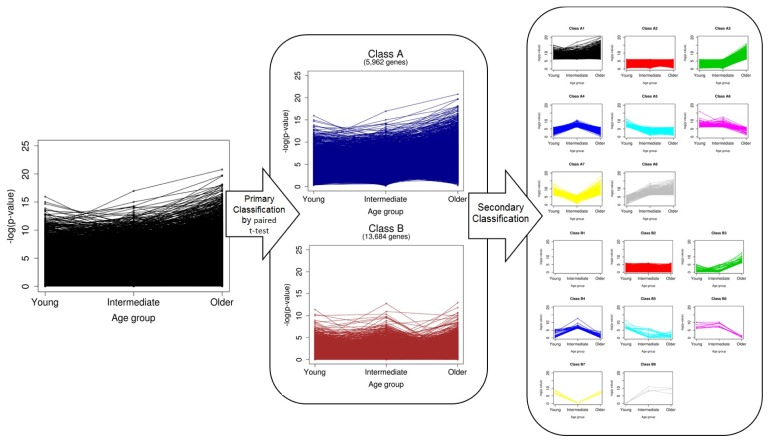
Transcriptome analysis has been widely used to make biomarker panels to diagnose cancers. In breast cancer, the age of the patient has been known to be associated with clinical features. As clinical transcriptome data have accumulated significantly, we classified all human genes based on age-specific differential expression between normal and breast cancer cells using public data. We retrieved the values for gene expression levels in breast cancer and matched normal cells from The Cancer Genome Atlas. We divided genes into two classes by paired t test without considering age in the first classification. We carried out a secondary classification of genes for each class into eight groups, based on the patterns of the p-values, which were calculated for each of the three age groups we defined. Through this two-step classification, gene expression was eventually grouped into 16 classes. We showed that this classification method could be applied to establish a more accurate prediction model to diagnose breast cancer by comparing the performance of prediction models with different combinations of genes. We expect that our scheme of classification could be used for other types of cancer data.
转录组分析已被广泛用于制作生物标志物面板以诊断癌症。在乳腺癌中,已知患者年龄与临床特征相关。随着临床转录组数据的大量积累,我们利用公开数据,根据正常细胞与乳腺癌细胞之间的年龄特异性差异表达对所有人类基因进行了分类。我们从癌症基因组图谱中获取了乳腺癌及匹配的正常细胞中基因表达水平的值。在第一次分类中,我们通过配对t检验将基因分为两类,不考虑年龄因素。基于为我们定义的三个年龄组各自计算的p值模式,我们对每个类别中的基因进行了二次分类,分为八组。通过这两步分类,基因表达最终被分为16类。我们表明,通过比较不同基因组合的预测模型的性能,这种分类方法可用于建立更准确的乳腺癌诊断预测模型。我们期望我们的分类方案可用于其他类型的癌症数据。