Center for Cancer Genome Discovery, DFCI, 450 Brookline Avenue, Dana840b, Boston, MA, 02215, USA.
Department of Pathology, Brigham and Women's Hospital, Boston, MA, USA.
BMC Genomics. 2018 Jan 8;19(1):30. doi: 10.1186/s12864-017-4428-5.
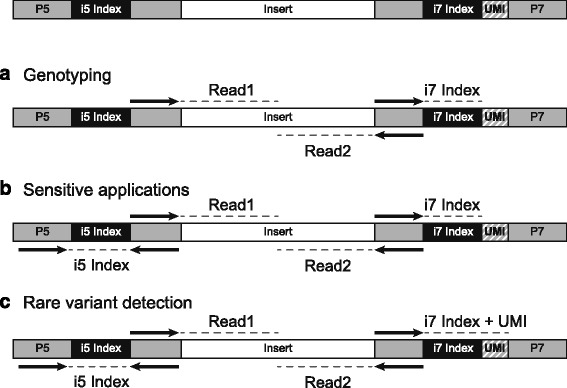
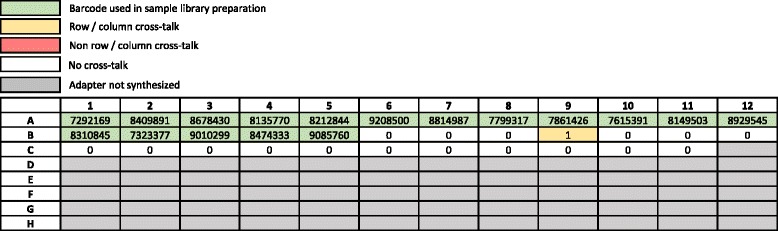
Sample index cross-talk can result in false positive calls when massively parallel sequencing (MPS) is used for sensitive applications such as low-frequency somatic variant discovery, ancient DNA investigations, microbial detection in human samples, or circulating cell-free tumor DNA (ctDNA) variant detection. Therefore, the limit-of-detection of an MPS assay is directly related to the degree of index cross-talk.
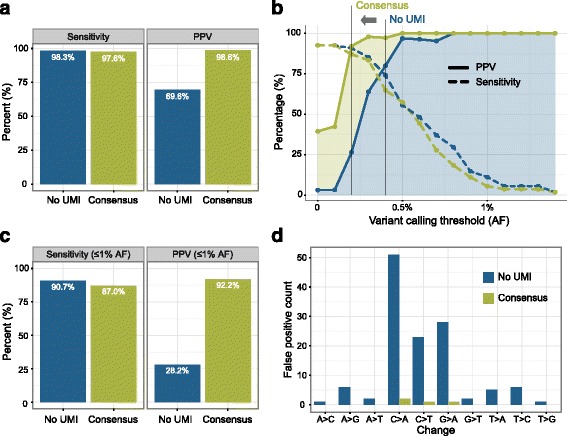
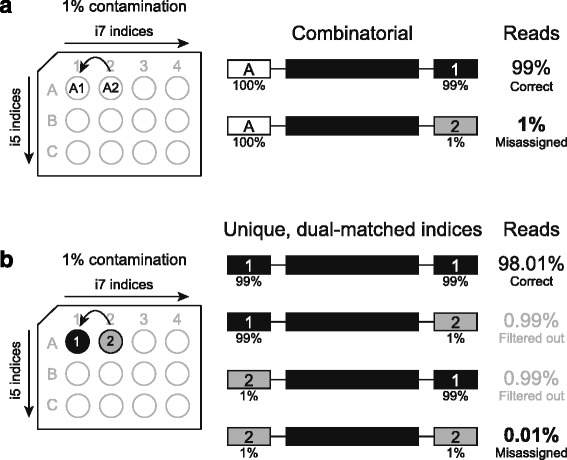
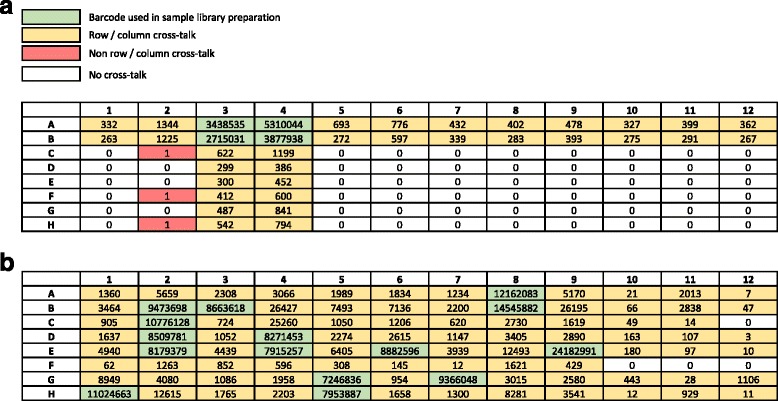
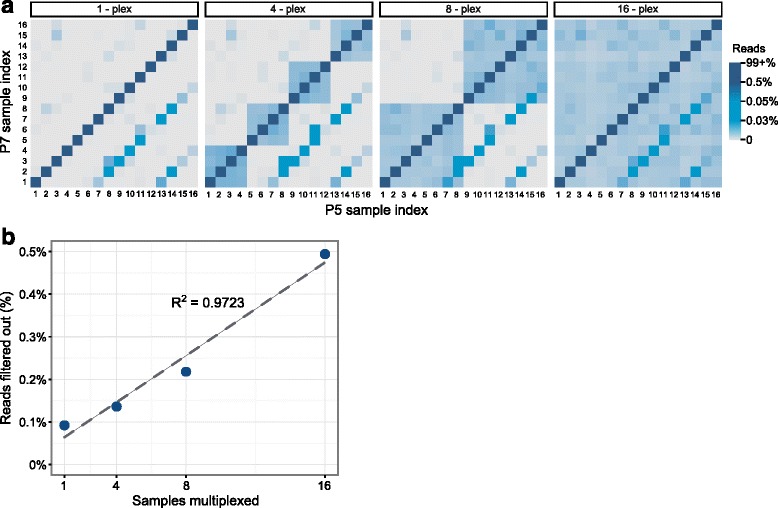
Cross-talk rates up to 0.29% were observed when using standard, combinatorial adapters, resulting in 110,180 (0.1% cross-talk rate) or 1,121,074 (0.29% cross-talk rate) misassigned reads per lane in non-patterned and patterned Illumina flow cells, respectively. Here, we demonstrate that using unique, dual-matched indexed adapters dramatically reduces index cross-talk to ≤1 misassigned reads per flow cell lane. While the current study was performed using dual-matched indices, using unique, dual-unrelated indices would also be an effective alternative.
For sensitive downstream analyses, the use of combinatorial indices for multiplexed hybrid capture and sequencing is inappropriate, as it results in an unacceptable number of misassigned reads. Cross-talk can be virtually eliminated using dual-matched indexed adapters. These results suggest that use of such adapters is critical to reduce false positive rates in assays that aim to identify low allele frequency events, and strongly indicate that dual-matched adapters be implemented for all sensitive MPS applications.
当大规模平行测序(MPS)用于敏感应用时,例如低频体细胞变异发现、古 DNA 研究、人类样本中的微生物检测或循环细胞游离肿瘤 DNA(ctDNA)变异检测,样品索引交叉干扰可能会导致假阳性结果。因此,MPS 检测的检测限直接与索引交叉干扰的程度有关。
当使用标准组合接头时,观察到的交叉干扰率高达 0.29%,导致非图案化和图案化 Illumina 流动细胞中每条泳道分别有 110,180(0.1%交叉干扰率)或 1,121,074(0.29%交叉干扰率)的错误分配读取。在这里,我们证明使用独特的、双匹配索引接头可显著降低索引交叉干扰,使每条流动细胞泳道的错误分配读取数≤1。虽然本研究使用双匹配索引进行,但使用独特的、双不相关的索引也是一种有效的替代方法。
对于敏感的下游分析,组合索引用于多重杂交捕获和测序是不合适的,因为它会导致不可接受数量的错误分配读取。使用双匹配索引接头可以几乎消除交叉干扰。这些结果表明,在旨在识别低等位基因频率事件的检测中,使用这种接头对于降低假阳性率至关重要,并强烈表明所有敏感的 MPS 应用都应使用双匹配接头。