The Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Department of Biostatistics, Harvard T. H. Chan School of Public Health, Boston, MA, USA.
Nat Microbiol. 2018 Mar;3(3):337-346. doi: 10.1038/s41564-017-0089-z. Epub 2018 Jan 8.
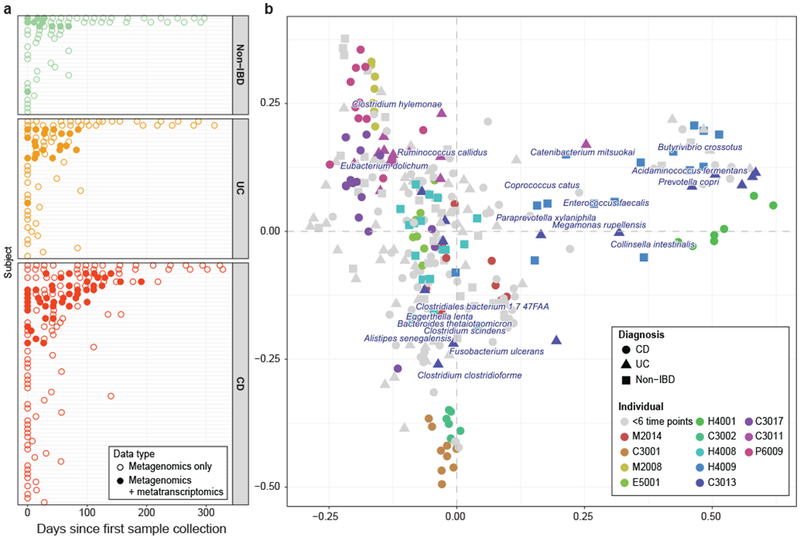
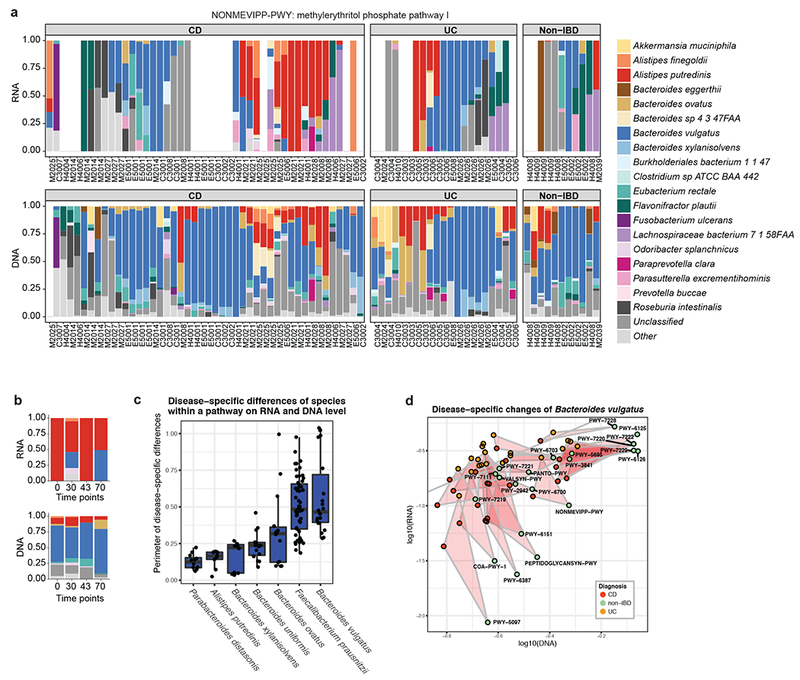
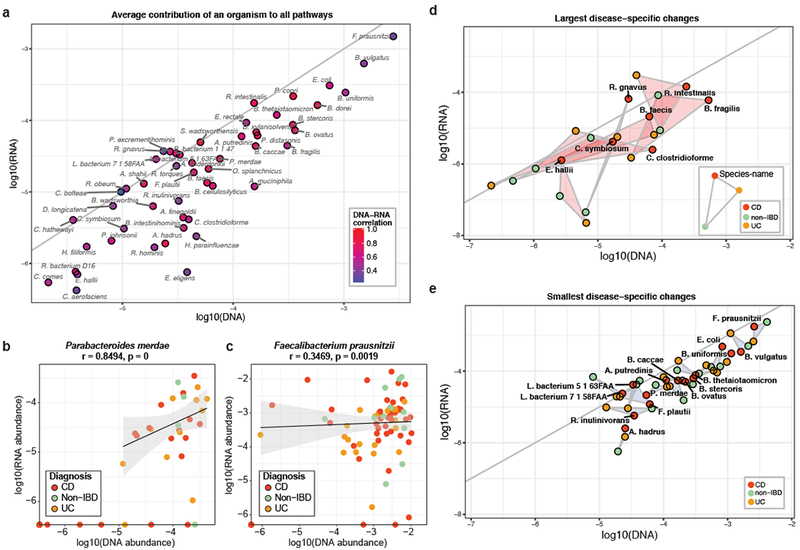
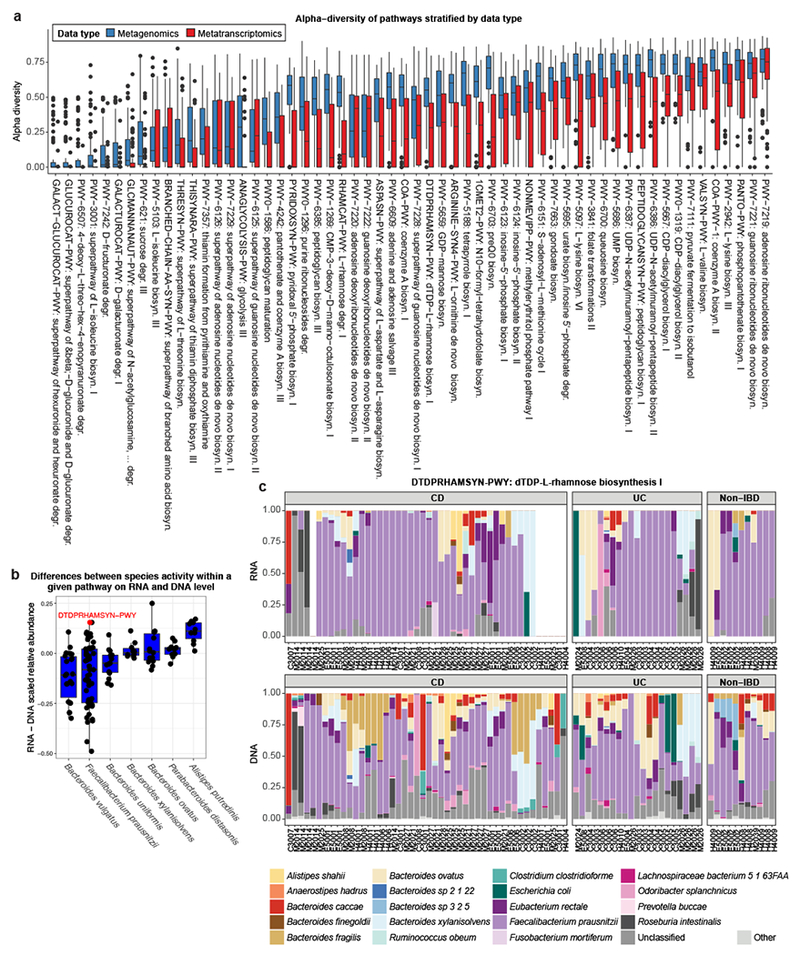
Inflammatory bowel disease (IBD) is a group of chronic diseases of the digestive tract that affects millions of people worldwide. Genetic, environmental and microbial factors have been implicated in the onset and exacerbation of IBD. However, the mechanisms associating gut microbial dysbioses and aberrant immune responses remain largely unknown. The integrative Human Microbiome Project seeks to close these gaps by examining the dynamics of microbiome functionality in disease by profiling the gut microbiomes of >100 individuals sampled over a 1-year period. Here, we present the first results based on 78 paired faecal metagenomes and metatranscriptomes, and 222 additional metagenomes from 59 patients with Crohn's disease, 34 with ulcerative colitis and 24 non-IBD control patients. We demonstrate several cases in which measures of microbial gene expression in the inflamed gut can be informative relative to metagenomic profiles of functional potential. First, although many microbial organisms exhibited concordant DNA and RNA abundances, we also detected species-specific biases in transcriptional activity, revealing predominant transcription of pathways by individual microorganisms per host (for example, by Faecalibacterium prausnitzii). Thus, a loss of these organisms in disease may have more far-reaching consequences than suggested by their genomic abundances. Furthermore, we identified organisms that were metagenomically abundant but inactive or dormant in the gut with little or no expression (for example, Dialister invisus). Last, certain disease-specific microbial characteristics were more pronounced or only detectable at the transcript level, such as pathways that were predominantly expressed by different organisms in patients with IBD (for example, Bacteroides vulgatus and Alistipes putredinis). This provides potential insights into gut microbial pathway transcription that can vary over time, inducing phenotypical changes that are complementary to those linked to metagenomic abundances. The study's results highlight the strength of analysing both the activity and the presence of gut microorganisms to provide insight into the role of the microbiome in IBD.
炎症性肠病(IBD)是一组影响全球数百万人的慢性消化道疾病。遗传、环境和微生物因素都与 IBD 的发病和恶化有关。然而,肠道微生物失调和异常免疫反应之间的关联机制在很大程度上仍不清楚。综合人类微生物组计划通过分析 100 多名个体在 1 年内采集的肠道微生物组,来研究微生物组功能在疾病中的动态,以此来填补这些空白。在这里,我们根据 78 对粪便宏基因组和宏转录组以及 59 名克罗恩病患者、34 名溃疡性结肠炎患者和 24 名非 IBD 对照患者的 222 个额外宏基因组,呈现了最初的研究结果。我们展示了一些案例,其中肠道炎症部位的微生物基因表达水平可以提供比功能潜力的宏基因组图谱更有价值的信息。首先,尽管许多微生物生物表现出 DNA 和 RNA 丰度的一致性,但我们也检测到转录活性的物种特异性偏差,揭示了每个宿主中单个微生物主导的途径转录(例如,普雷沃氏菌属)。因此,这些生物在疾病中的缺失可能比它们的基因组丰度所暗示的具有更深远的后果。此外,我们发现了一些在肠道中宏基因组丰富但活性低或休眠的微生物,其表达水平很低或没有(例如,隐匿真杆菌)。最后,某些特定于疾病的微生物特征在转录水平上更加明显或仅可检测到,例如在 IBD 患者中主要由不同微生物表达的途径(例如,脆弱拟杆菌和腐败真杆菌)。这为肠道微生物途径转录提供了潜在的见解,这些转录可能随时间而变化,诱导表型变化,与与宏基因组丰度相关的变化互补。该研究的结果强调了分析肠道微生物的活性和存在以深入了解微生物组在 IBD 中的作用的优势。