Huang Kai-Yao, Chang Tzu-Hao, Jhong Jhih-Hua, Chi Yu-Hsiang, Li Wen-Chi, Chan Chien-Lung, Robert Lai K, Lee Tzong-Yi
Department of Computer Science and Engineering, Yuan Ze University, Taoyuan City, 320, Taiwan.
Department of Medical Research, Hsinchu Mackay Memorial Hospital, Hsinchu City, 300, Taiwan.
BMC Syst Biol. 2017 Dec 21;11(Suppl 7):131. doi: 10.1186/s12918-017-0503-4.
Anti-microbial peptides (AMPs), naturally encoded by genes and generally containing 12-100 amino acids, are crucial components of the innate immune system and can protect the host from various pathogenic bacteria and viruses. In recent years, the widespread use of antibiotics has resulted in the rapid growth of antibiotic-resistant microorganisms that often induce critical infection and pathogenesis. Recently, the advent of high-throughput technologies has led molecular biology into a data surge in both the amount and scope of data. For instance, next-generation sequencing technology has been applied to generate large-scale sequencing reads from foods, water, soil, air, and specimens to identify microbiota and their functions based on metagenomics and metatranscriptomics, respectively. In addition, oolong tea is partially fermented and is the most widely produced tea in Taiwan. Many studies have shown the benefits of oolong tea in inhibiting obesity, reducing dental plaque deposition, antagonizing allergic immune responses, and alleviating the effects of aging. However, the microbes and their functions present in oolong tea remain unknown.
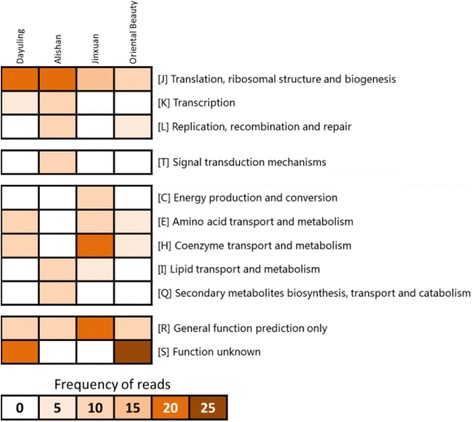
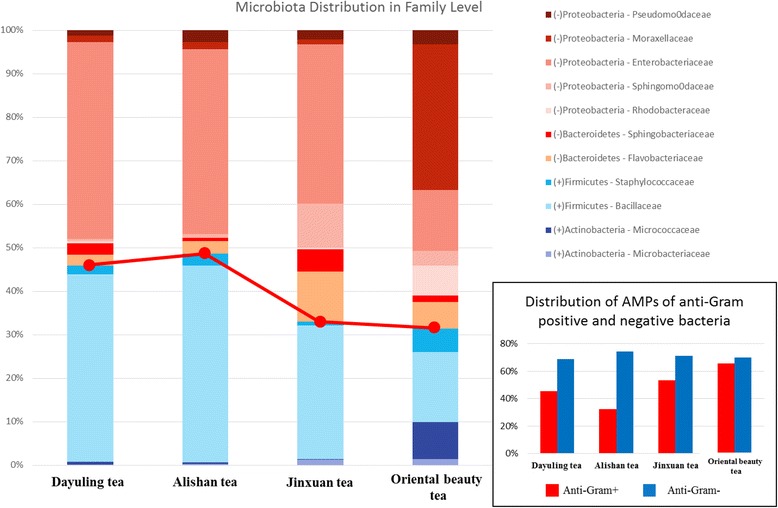
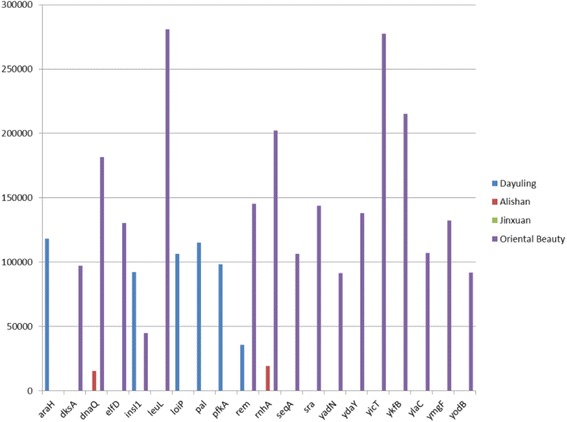
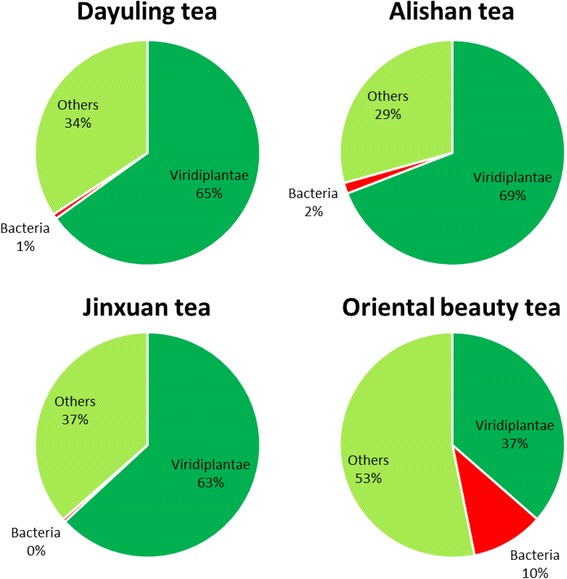
To understand the relationship between Taiwanese oolong teas and bacterial communities, we designed a novel bioinformatics scheme to identify AMPs and their functional types based on metagenomics and metatranscriptomic analysis of high-throughput transcriptome data. Four types of oolong teas (Dayuling tea, Alishan tea, Jinxuan tea, and Oriental Beauty tea) were subjected to 16S ribosomal DNA and total RNA extraction and sequencing. Metagenomics analysis results revealed that Oriental Beauty tea exhibited greater bacterial diversity than other teas. The most common bacterial families across all tea types were Bacteroidaceae (21.7%), Veillonellaceae (22%), and Fusobacteriaceae (12.3%). Metatranscriptomics analysis results revealed that the dominant bacteria species across all tea types were Escherichia coli, Bacillus subtilis, and Chryseobacterium sp. StRB126, which were subjected to further functional analysis. A total of 8194 (6.5%), 26,220 (6.1%), 5703 (5.8%), and 106,183 (7.8%) reads could be mapped to AMPs.
We found that the distribution of anti-gram-positive and anti-gram-negative AMPs is highly correlated with the distribution of gram-positive and gram-negative bacteria in Taiwanese oolong tea samples.
抗菌肽(AMPs)由基因天然编码,通常含有12 - 100个氨基酸,是先天免疫系统的关键组成部分,可保护宿主免受各种病原菌和病毒的侵害。近年来,抗生素的广泛使用导致了抗生素耐药微生物的迅速增长,这些微生物常常引发严重感染和发病机制。最近,高通量技术的出现使分子生物学在数据量和数据范围上都进入了一个数据激增的阶段。例如,下一代测序技术已被应用于从食物、水、土壤、空气和标本中生成大规模测序读数,分别基于宏基因组学和宏转录组学来鉴定微生物群及其功能。此外,乌龙茶是半发酵茶,是台湾产量最高的茶叶。许多研究表明乌龙茶在抑制肥胖、减少牙菌斑沉积、拮抗过敏免疫反应和缓解衰老影响方面具有益处。然而,乌龙茶中存在的微生物及其功能仍然未知。
为了了解台湾乌龙茶与细菌群落之间的关系,我们设计了一种新颖的生物信息学方案,基于高通量转录组数据的宏基因组学和宏转录组学分析来鉴定抗菌肽及其功能类型。对四种乌龙茶(大禹岭茶、阿里山茶、金萱茶和东方美人茶)进行了16S核糖体DNA和总RNA提取及测序。宏基因组学分析结果显示,东方美人茶的细菌多样性高于其他茶叶。所有茶类中最常见的细菌科是拟杆菌科(21.7%)、韦荣球菌科(22%)和梭杆菌科(12.3%)。宏转录组学分析结果显示,所有茶类中的优势细菌种类是大肠杆菌、枯草芽孢杆菌和金黄杆菌属StRB126,并对其进行了进一步的功能分析。共有8194条(6.5%)、26220条(6.1%)、5703条(5.8%)和106183条(7.8%)读数可映射到抗菌肽。
我们发现台湾乌龙茶样品中抗革兰氏阳性和抗革兰氏阴性抗菌肽的分布与革兰氏阳性和革兰氏阴性细菌的分布高度相关。