Dayton Jonathan B, Piccolo Stephen R
Department of Biology, Brigham Young University, Provo, UT, 84602, USA.
Department of Biomedical Informatics, University of Utah, Salt Lake City, UT, 84108, USA.
BMC Med Genomics. 2017 Dec 21;10(Suppl 4):66. doi: 10.1186/s12920-017-0303-0.
Malignant tumors are typically caused by a conglomeration of genomic aberrations-including point mutations, small insertions, small deletions, and large copy-number variations. In some cases, specific chemotherapies and targeted drug treatments are effective against tumors that harbor certain genomic aberrations. However, predictive aberrations (biomarkers) have not been identified for many tumor types and treatments. One way to address this problem is to examine the downstream, transcriptional effects of genomic aberrations and to identify characteristic patterns. Even though two tumors harbor different genomic aberrations, the transcriptional effects of those aberrations may be similar. These patterns could be used to inform treatment choices.
We used data from 9300 tumors across 25 cancer types from The Cancer Genome Atlas. We used supervised machine learning to evaluate our ability to distinguish between tumors that had mutually exclusive genomic aberrations in specific genes. An ability to accurately distinguish between tumors with aberrations in these genes suggested that the genes have a relatively different downstream effect on transcription, and vice versa. We compared these findings against prior knowledge about signaling networks and drug responses.
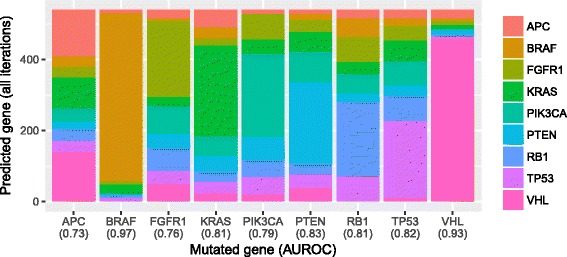
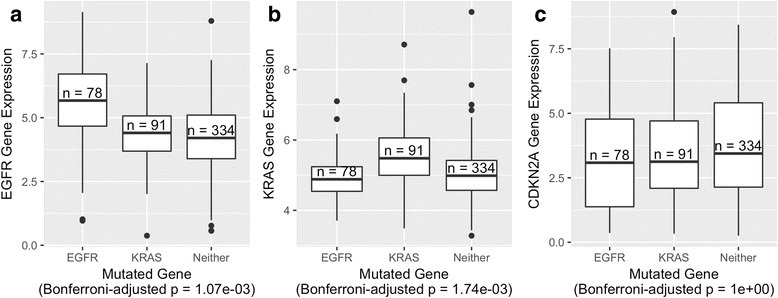
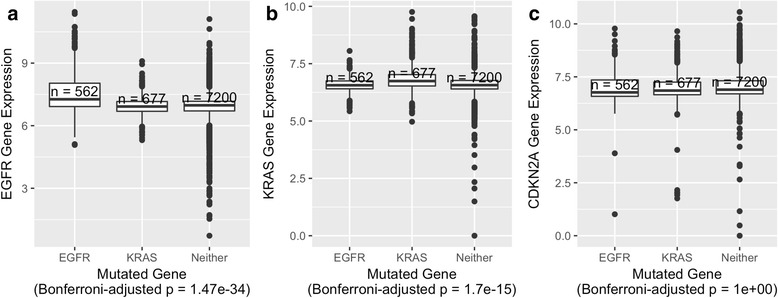
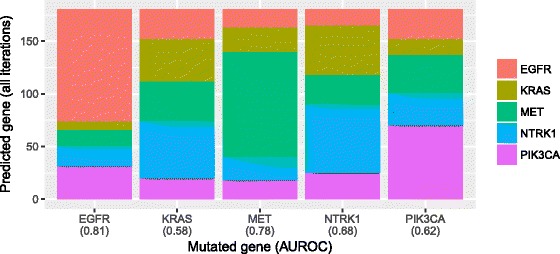
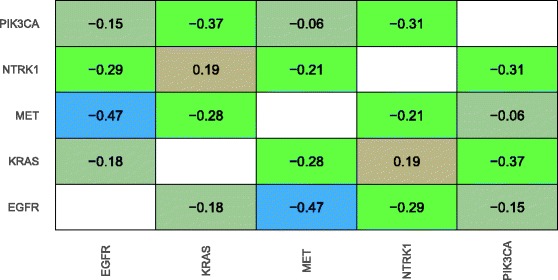
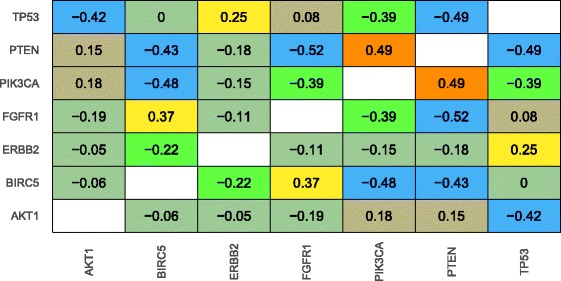
Our analysis recapitulates known relationships in cancer pathways and identifies gene pairs known to predict responses to the same treatments. For example, in lung adenocarcinomas, gene-expression profiles from tumors with somatic aberrations in EGFR or MET were negatively correlated with each other, in line with prior knowledge that MET amplification causes resistance to EGFR inhibition. In breast carcinomas, we observed high similarity between PTEN and PIK3CA, which play complementary roles in regulating cellular proliferation. In a pan-cancer analysis, we found that genomic aberrations in BRAF and VHL exhibit downstream effects that are clearly distinct from other genes.
We show that transcriptional data offer promise as a way to group genomic aberrations according to their downstream effects, and these groupings recapitulate known relationships. Our approach shows potential to help pharmacologists and clinical trialists narrow the search space for candidate gene/drug associations, including for rare mutations, and for identifying potential drug-repurposing opportunities.
恶性肿瘤通常由基因组畸变聚集引起,包括点突变、小插入、小缺失和大拷贝数变异。在某些情况下,特定的化疗和靶向药物治疗对具有某些基因组畸变的肿瘤有效。然而,许多肿瘤类型和治疗方法尚未确定预测性畸变(生物标志物)。解决这一问题的一种方法是研究基因组畸变的下游转录效应,并识别特征模式。即使两个肿瘤具有不同的基因组畸变,这些畸变的转录效应可能相似。这些模式可用于指导治疗选择。
我们使用了来自癌症基因组图谱中25种癌症类型的9300个肿瘤的数据。我们使用监督机器学习来评估我们区分特定基因中具有互斥基因组畸变的肿瘤的能力。准确区分这些基因中存在畸变的肿瘤的能力表明这些基因对转录具有相对不同的下游效应,反之亦然。我们将这些发现与关于信号网络和药物反应的现有知识进行了比较。
我们的分析概括了癌症通路中的已知关系,并识别了已知可预测对相同治疗反应的基因对。例如,在肺腺癌中,表皮生长因子受体(EGFR)或间质-上皮转化因子(MET)存在体细胞畸变的肿瘤的基因表达谱彼此呈负相关,这与MET扩增导致对EGFR抑制产生耐药性的现有知识一致。在乳腺癌中,我们观察到在调节细胞增殖中起互补作用的磷酸酶及张力蛋白同源物(PTEN)和磷脂酰肌醇-3激酶催化亚基α(PIK3CA)之间具有高度相似性。在泛癌分析中,我们发现BRAF和VHL的基因组畸变表现出与其他基因明显不同的下游效应。
我们表明转录数据有望作为一种根据基因组畸变的下游效应将其分组的方法,并且这些分组概括了已知关系。我们的方法显示出帮助药理学家和临床试验人员缩小候选基因/药物关联搜索空间的潜力,包括针对罕见突变,以及识别潜在的药物重新利用机会。