Lu Qing, Niu Xiaojun, Zhang Mengchen, Wang Caihong, Xu Qun, Feng Yue, Yang Yaolong, Wang Shan, Yuan Xiaoping, Yu Hanyong, Wang Yiping, Chen Xiaoping, Liang Xuanqiang, Wei Xinghua
State Key Laboratory of Rice Biology, China National Rice Research Institute, Hangzhou, China.
Crops Research Institute, Guangdong Academy of Agricultural Sciences, South China Peanut Sub-Center of National Center of Oilseed Crops Improvement and Guangdong Provincial Key Laboratory of Crop Genetic Improvement, Guangzhou, China.
Front Plant Sci. 2018 Jan 5;8:2213. doi: 10.3389/fpls.2017.02213. eCollection 2017.
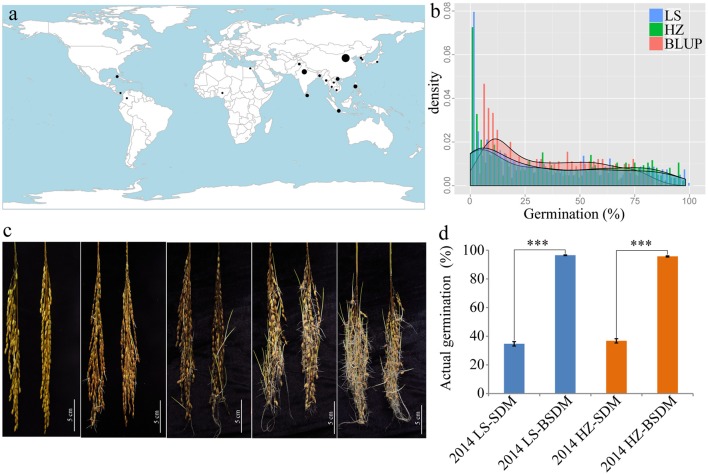
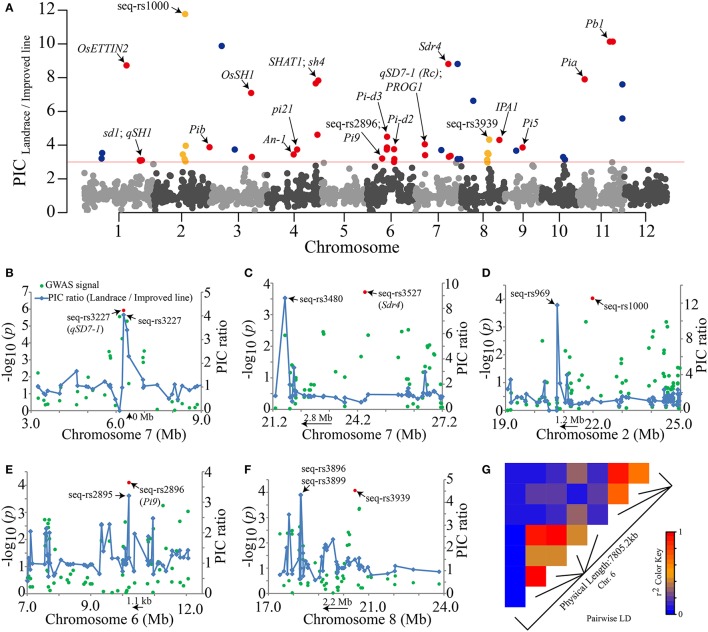
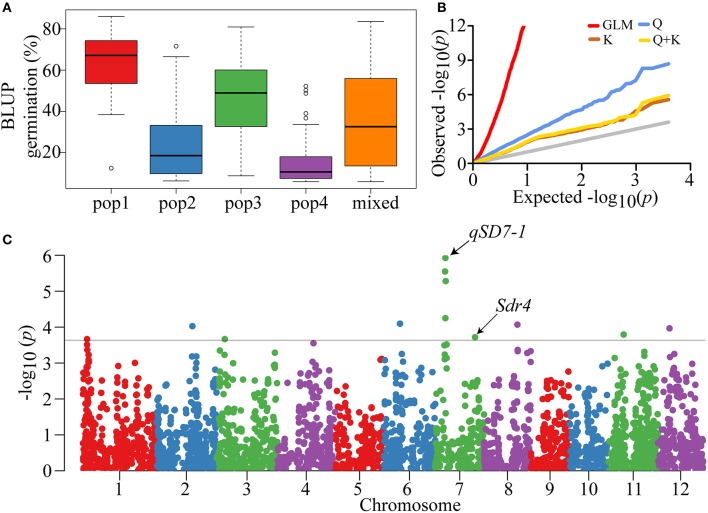
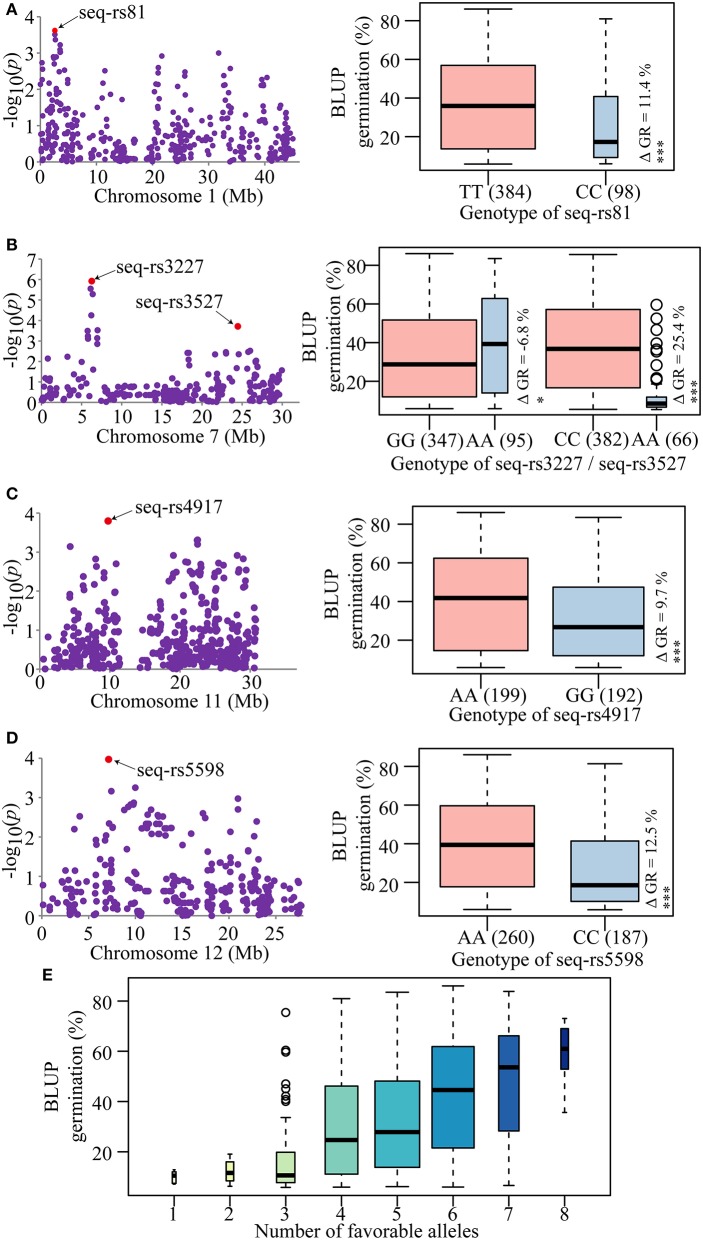
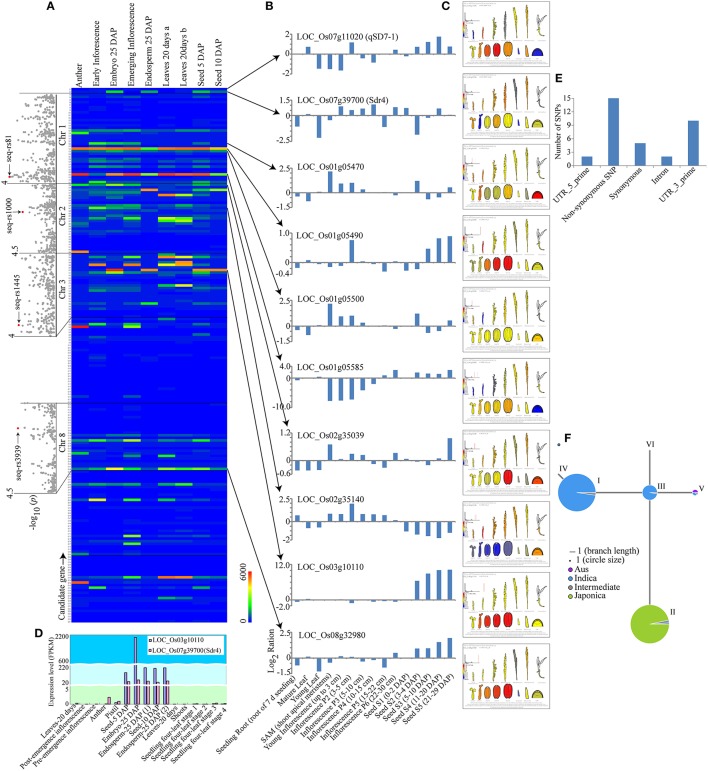
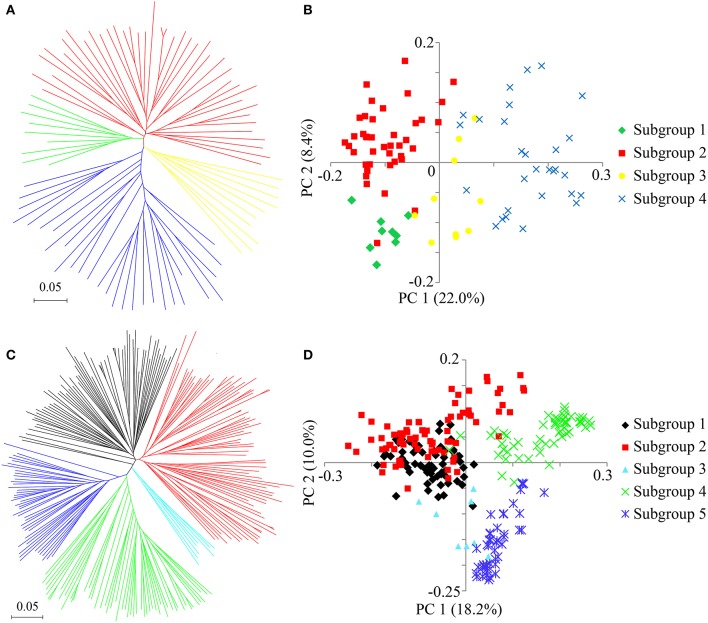
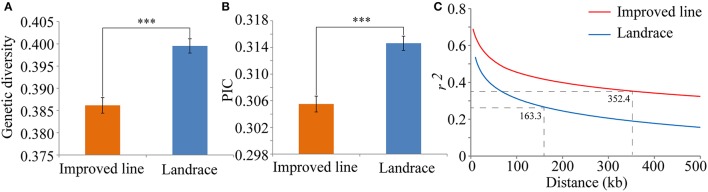
Seed dormancy is an important agronomic trait affecting grain yield and quality because of pre-harvest germination and is influenced by both environmental and genetic factors. However, our knowledge of the factors controlling seed dormancy remains limited. To better reveal the molecular mechanism underlying this trait, a genome-wide association study was conducted in an -only population consisting of 453 accessions genotyped using 5,291 SNPs. Nine known and new significant SNPs were identified on eight chromosomes. These lead SNPs explained 34.9% of the phenotypic variation, and four of them were designed as dCAPS markers in the hope of accelerating molecular breeding. Moreover, a total of 212 candidate genes was predicted and eight candidate genes showed plant tissue-specific expression in expression profile data from different public bioinformatics databases. In particular, LOC_Os03g10110, which had a maize homolog involved in embryo development, was identified as a candidate regulator for further biological function investigations. Additionally, a polymorphism information content ratio method was used to screen improvement footprints and 27 selective sweeps were identified, most of which harbored domestication-related genes. Further studies suggested that three significant SNPs were adjacent to the candidate selection signals, supporting the accuracy of our genome-wide association study (GWAS) results. These findings show that genome-wide screening for selective sweeps can be used to identify new improvement-related DNA regions, although the phenotypes are unknown. This study enhances our knowledge of the genetic variation in seed dormancy, and the new dormancy-associated SNPs will provide real benefits in molecular breeding.
种子休眠是一个重要的农艺性状,由于收获前发芽会影响谷物产量和品质,并且受环境和遗传因素的影响。然而,我们对控制种子休眠的因素的了解仍然有限。为了更好地揭示这一性状的分子机制,在一个由453份材料组成的仅含一个群体中进行了全基因组关联研究,这些材料使用5291个单核苷酸多态性(SNP)进行了基因分型。在8条染色体上鉴定出9个已知的和新的显著SNP。这些主效SNP解释了34.9%的表型变异,其中4个被设计为dCAPS标记,以期加速分子育种。此外,共预测了212个候选基因,8个候选基因在来自不同公共生物信息学数据库的表达谱数据中表现出植物组织特异性表达。特别是,LOC_Os03g10110有一个参与胚胎发育的玉米同源基因被鉴定为候选调控因子,用于进一步的生物学功能研究。此外,使用多态性信息含量比方法筛选改良足迹,鉴定出27个选择性清除区域,其中大多数含有与驯化相关的基因。进一步的研究表明,3个显著SNP与候选选择信号相邻,支持了我们全基因组关联研究(GWAS)结果的准确性。这些发现表明,尽管表型未知,但对选择性清除区域进行全基因组筛选可用于鉴定与改良相关的新DNA区域。本研究增进了我们对种子休眠遗传变异的了解,新的与休眠相关的SNP将在分子育种中带来实际益处。