Mortimer Tatum D, Weber Alexandra M, Pepperell Caitlin S
Division of Infectious Diseases, Department of Medicine, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
mSystems. 2018 Jan 30;3(1). doi: 10.1128/mSystems.00108-17. eCollection 2018 Jan-Feb.
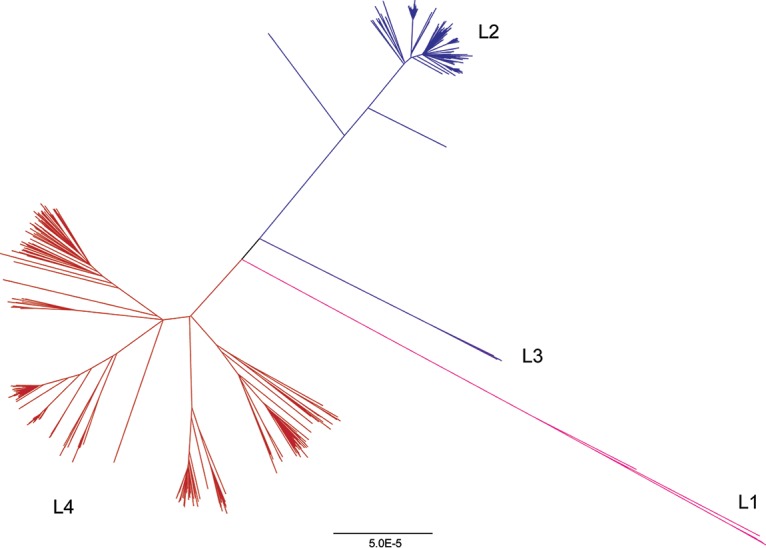
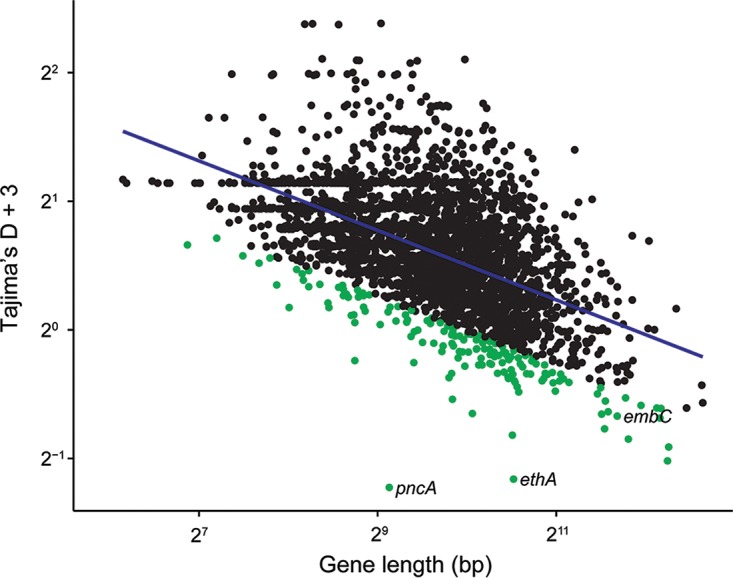
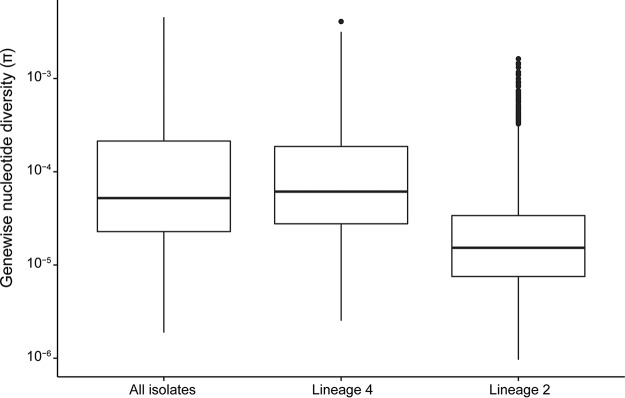
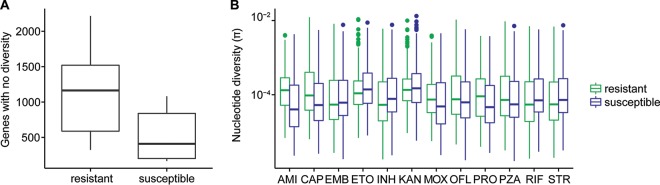
Tuberculosis (TB) is the leading cause of death by an infectious disease, and global TB control efforts are increasingly threatened by drug resistance in . Unlike most bacteria, where lateral gene transfer is an important mechanism of resistance acquisition, resistant arises solely by chromosomal mutation. Using whole-genome sequencing data from two natural populations of , we characterized the population genetics of known drug resistance loci using measures of diversity, population differentiation, and convergent evolution. We found resistant subpopulations to be less diverse than susceptible subpopulations, consistent with ongoing transmission of resistant . A subset of resistance genes ("sloppy targets") were characterized by high diversity and multiple rare variants; we posit that a large genetic target for resistance and relaxation of purifying selection contribute to high diversity at these loci. For "tight targets" of selection, the path to resistance appeared narrower, evidenced by single favored mutations that arose numerous times in the phylogeny and segregated at markedly different frequencies in resistant and susceptible subpopulations. These results suggest that diverse genetic architectures underlie drug resistance in and that combined approaches are needed to identify causal mutations. Extrapolating from patterns observed for well-characterized genes, we identified novel candidate variants involved in resistance. The approach outlined here can be extended to identify resistance variants for new drugs, to investigate the genetic architecture of resistance, and when phenotypic data are available, to find candidate genetic loci underlying other positively selected traits in clonal bacteria. , the causative agent of tuberculosis (TB), is a significant burden on global health. Antibiotic treatment imposes strong selective pressure on populations. Identifying the mutations that cause drug resistance in is important for guiding TB treatment and halting the spread of drug resistance. Whole-genome sequencing (WGS) of isolates can be used to identify novel mutations mediating drug resistance and to predict resistance patterns faster than traditional methods of drug susceptibility testing. We have used WGS from natural populations of drug-resistant to characterize effects of selection for advantageous mutations on patterns of diversity at genes involved in drug resistance. The methods developed here can be used to identify novel advantageous mutations, including new resistance loci, in and other clonal pathogens.
结核病(TB)是传染病致死的首要原因,全球结核病控制工作日益受到耐药性的威胁。与大多数细菌不同,横向基因转移是其获得耐药性的重要机制,而耐药结核杆菌仅通过染色体突变产生。利用来自两个结核杆菌自然种群的全基因组测序数据,我们通过多样性、种群分化和趋同进化等指标,对已知耐药位点的种群遗传学特征进行了描述。我们发现耐药亚群的多样性低于敏感亚群,这与耐药结核杆菌的持续传播相一致。一部分耐药基因(“宽松靶点”)具有高多样性和多个罕见变异;我们推测,耐药的大遗传靶点和纯化选择的放松导致了这些位点的高多样性。对于选择的“紧密靶点”,耐药途径似乎更窄,系统发育中多次出现单一优势突变,且在耐药和敏感亚群中的分离频率明显不同,这证明了这一点。这些结果表明,多种遗传结构是结核杆菌耐药性的基础,需要综合方法来识别因果突变。从特征明确的基因所观察到的模式进行推断,我们鉴定出了涉及耐药性的新候选变异。这里概述的方法可以扩展到识别新药的耐药变异,研究耐药性的遗传结构,以及在有表型数据时,寻找克隆细菌中其他正选择性状的潜在候选基因座。结核分枝杆菌是结核病(TB)的病原体,对全球健康造成重大负担。抗生素治疗对结核分枝杆菌种群施加了强大的选择压力。识别导致结核分枝杆菌耐药性的突变对于指导结核病治疗和阻止耐药性传播至关重要。结核分枝杆菌分离株的全基因组测序(WGS)可用于识别介导耐药性的新突变,并比传统药敏试验方法更快地预测耐药模式。我们利用耐药结核分枝杆菌自然种群的WGS来描述有利突变选择对耐药相关基因多样性模式的影响。这里开发的方法可用于识别结核分枝杆菌和其他克隆病原体中的新的有利突变,包括新的耐药位点。