Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of California, San Francisco, CA, USA.
Department of Medicine, Vanderbilt University, Nashville, TN, USA.
Lancet Respir Med. 2018 Feb;6(2):154-160. doi: 10.1016/S2213-2600(18)30007-9.
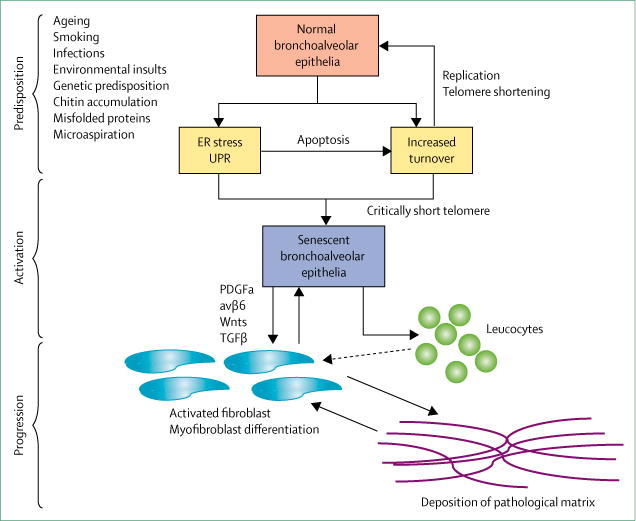
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and typically fatal lung disease characterised by subpleural fibrosis, subepithelial fibroblast foci, and microscopic honeycombing. Although understanding of the pathogenic mechanisms continues to evolve, evidence indicates that distal airway and alveolar epithelial cells are central drivers of the disease. In this Viewpoint, we review the history of naming and classifications used to define the disease now referred to as IPF, in the context of understanding the clinical presentation, causes, and pathogenesis of the disease. We aim to generate discussion on whether, given the substantial progress made in understanding the clinical, genetic, cellular, and molecular mechanisms involved in the development of IPF, a change of name should be considered. To initiate this discussion, we offer new suggestions to update the name of this disease and new approaches to classify all forms of pulmonary fibrosis.
特发性肺纤维化(IPF)是一种进行性、不可逆转且通常致命的肺部疾病,其特征为亚胸膜纤维化、上皮下成纤维细胞灶和微观蜂窝状改变。尽管对发病机制的理解仍在不断发展,但有证据表明,远端气道和肺泡上皮细胞是该疾病的主要驱动因素。在本观点中,我们回顾了用于定义目前称为 IPF 的疾病的命名和分类的历史,以期了解疾病的临床表现、病因和发病机制。我们旨在讨论是否应该考虑更改名称,鉴于在理解 IPF 发生的临床、遗传、细胞和分子机制方面取得了重大进展。为了启动这一讨论,我们提出了更新该疾病名称的新建议和对所有类型的肺纤维化进行分类的新方法。