a Heart and Kidney Institute, College of Pharmacy , University of Houston , Houston , TX , USA.
Clin Exp Hypertens. 2019;41(1):5-13. doi: 10.1080/10641963.2018.1433197. Epub 2018 Feb 9.
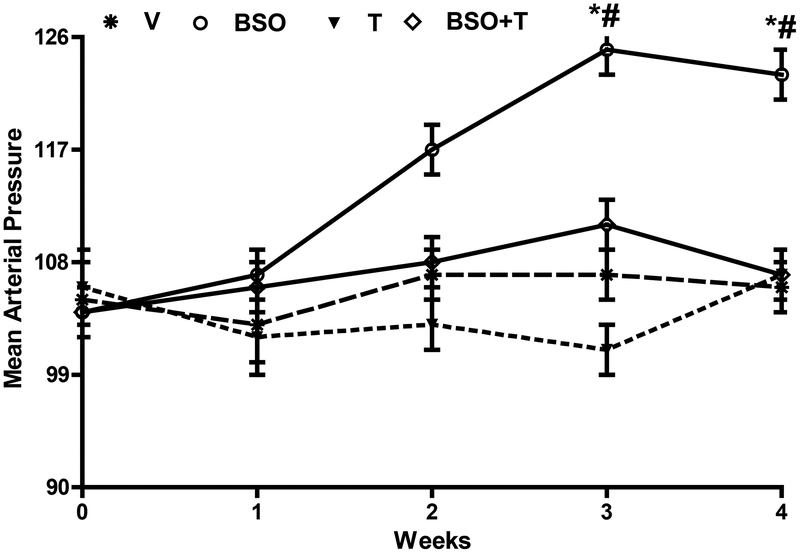
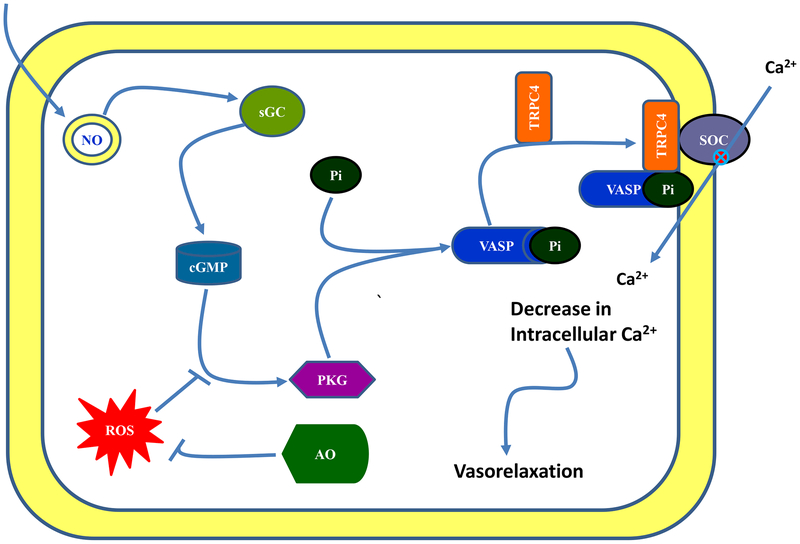
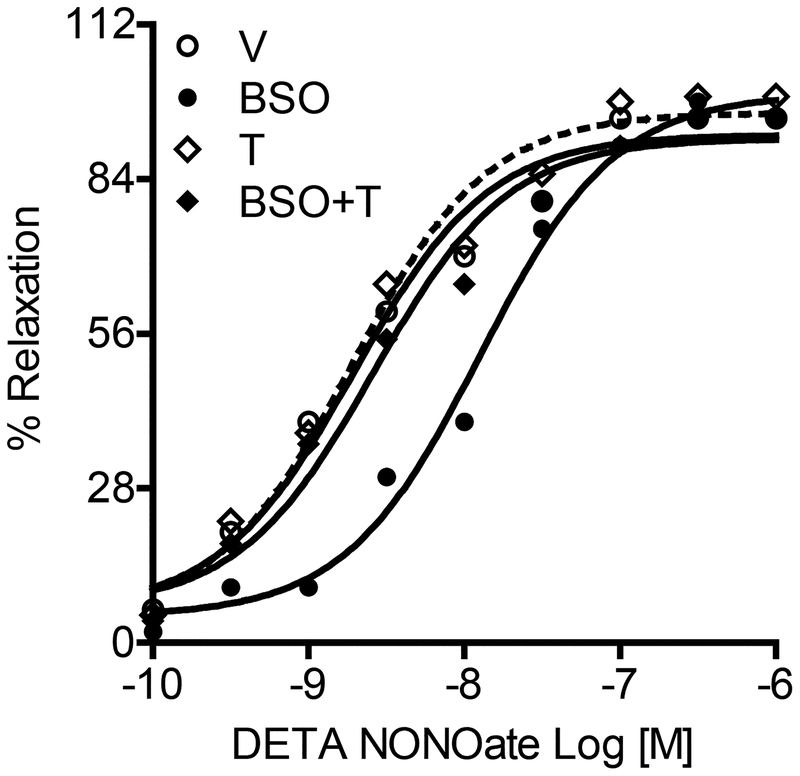
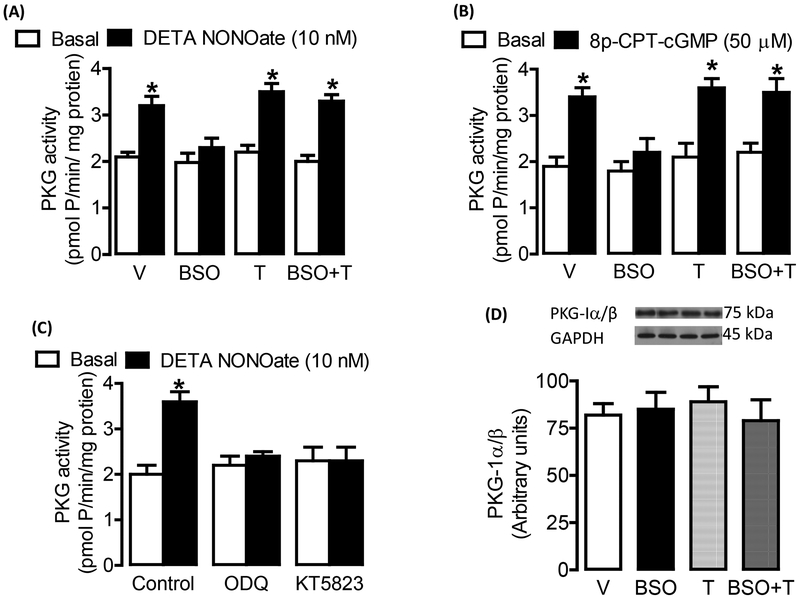
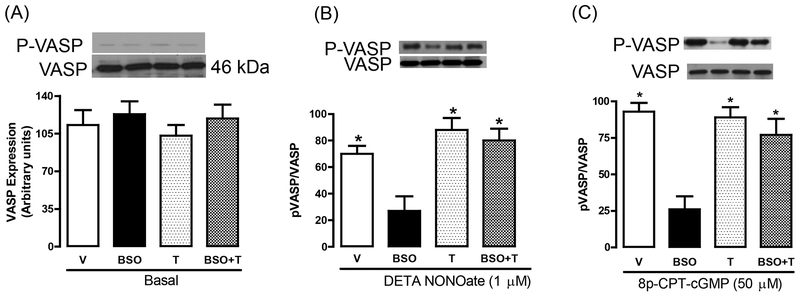
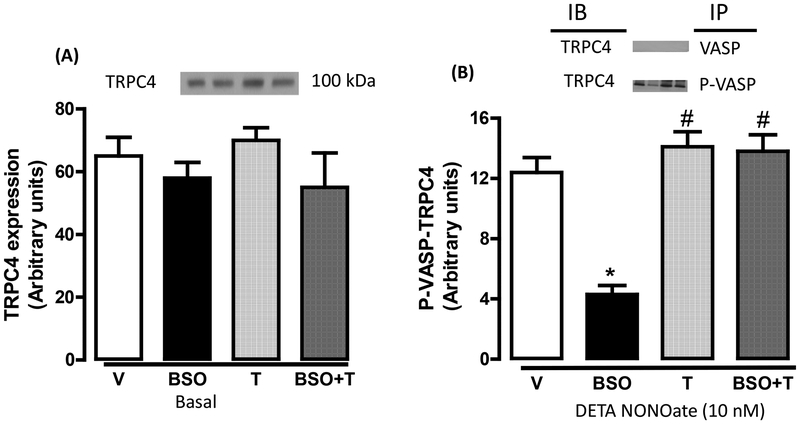
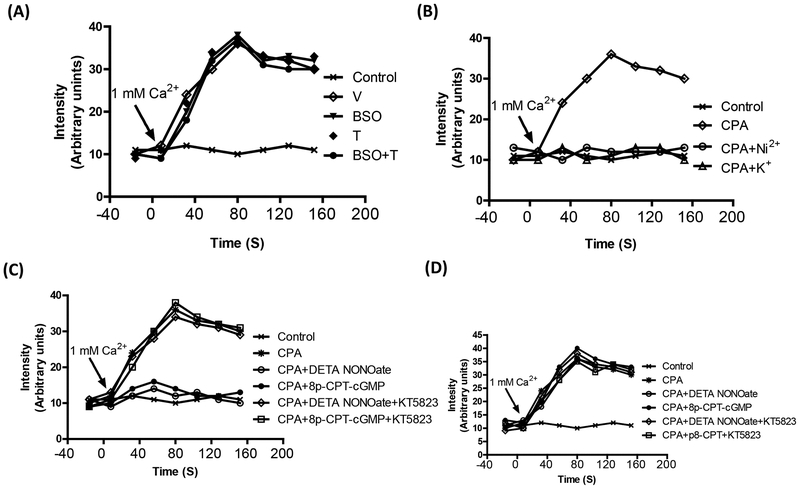
Reactive oxygen species induce vascular dysfunction and hypertension by directly interacting with nitric oxide (NO) which leads to NO inactivation. In addition to a decrease in NO bioavailability, there is evidence that oxidative stress can also modulate NO signaling during hypertension. Here, we investigated the effect of oxidative stress on NO signaling molecules cGMP-dependent protein kinase (PKG) and vasodilator-stimulated phosphoprotein (VASP) which are known to mediate vasodilatory actions of NO. Male Sprague Dawley (SD) rats were provided with tap water (control), 30 mM L-buthionine sulfoximine (BSO, a pro-oxidant), 1 mM tempol (T, an antioxidant) and BSO + T for 3 wks. BSO-treated rats exhibited high blood pressure and oxidative stress. Incubation of mesenteric arterial rings with NO donors caused concentration-dependent relaxation in control rats. However, the response to NO donors was significantly lower in BSO-treated rats with a marked decrease in pD2. In control rats, NO donors activated mesenteric PKG, increased VASP phosphorylation and its interaction with transient receptor potential channels 4 (TRPC4) and inhibited store-operated Ca influx. NO failed to activate these signaling molecules in mesenteric arteries from BSO-treated rats. Supplementation of BSO-treated rats with tempol reduced oxidative stress and blood pressure and normalized the NO signaling. These data suggest that oxidative stress can reduce NO-mediated PKG activation and VASP-TRPC4 interaction which leads to failure of NO to reduce Ca influx in smooth muscle cells. The increase in intracellular Ca contributes to sustained vasoconstriction and subsequent hypertension. Antioxidant supplementation decreases oxidative stress, normalizes NO signaling and reduces blood pressure.
活性氧自由基(ROS)通过与一氧化氮(NO)直接相互作用导致 NO 失活,从而引起血管功能障碍和高血压。除了降低 NO 的生物利用度外,有证据表明氧化应激也可以在高血压期间调节 NO 信号。在这里,我们研究了氧化应激对 NO 信号分子环磷酸鸟苷(cGMP)依赖性蛋白激酶(PKG)和血管扩张刺激磷蛋白(VASP)的影响,这两种分子已知介导 NO 的血管舒张作用。雄性 Sprague Dawley(SD)大鼠给予自来水(对照)、30mM L-丁硫氨酸亚砜(BSO,一种促氧化剂)、1mM 四甲基哌啶氧化物(T,一种抗氧化剂)和 BSO+T 共 3 周。BSO 处理的大鼠表现出高血压和氧化应激。用 NO 供体孵育肠系膜动脉环,在对照大鼠中引起浓度依赖性松弛。然而,NO 供体对 BSO 处理的大鼠的反应明显降低,pD2 明显降低。在对照大鼠中,NO 供体激活肠系膜 PKG,增加 VASP 磷酸化及其与瞬时受体电位通道 4(TRPC4)的相互作用,并抑制储存操作的 Ca 内流。NO 不能激活 BSO 处理的大鼠肠系膜动脉中的这些信号分子。用 TEMPOL 补充 BSO 处理的大鼠可降低氧化应激和血压,并使 NO 信号正常化。这些数据表明,氧化应激可以降低 NO 介导的 PKG 激活和 VASP-TRPC4 相互作用,导致 NO 不能减少平滑肌细胞中的 Ca 内流。细胞内 Ca 的增加导致持续的血管收缩和随后的高血压。抗氧化剂补充可降低氧化应激、使 NO 信号正常化并降低血压。