Hoffmann Thorsten M, Cwiklinski Emma, Shah Dinesh S, Stretton Clare, Hyde Russell, Taylor Peter M, Hundal Harinder S
Division of Cell Signalling and Immunology, Sir James Black Centre, School of Life Sciences, University of Dundee, Dundee, United Kingdom.
Front Pharmacol. 2018 Feb 7;9:63. doi: 10.3389/fphar.2018.00063. eCollection 2018.
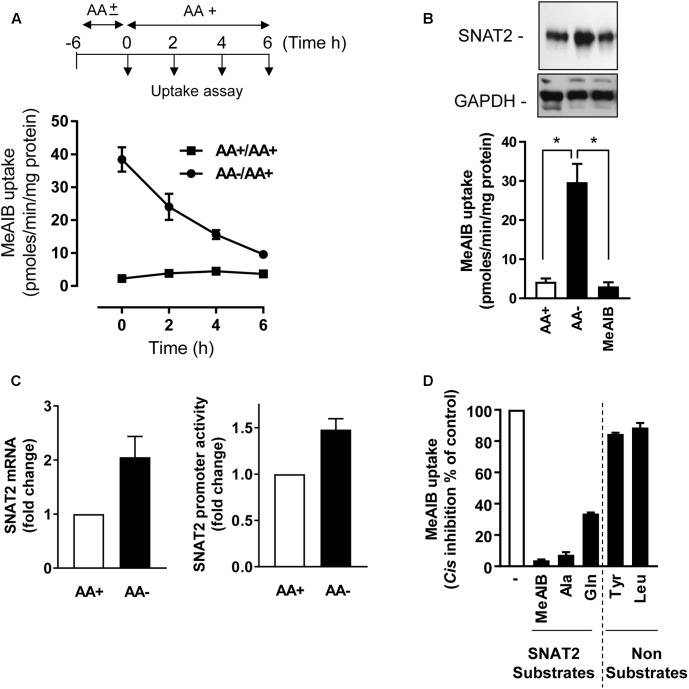
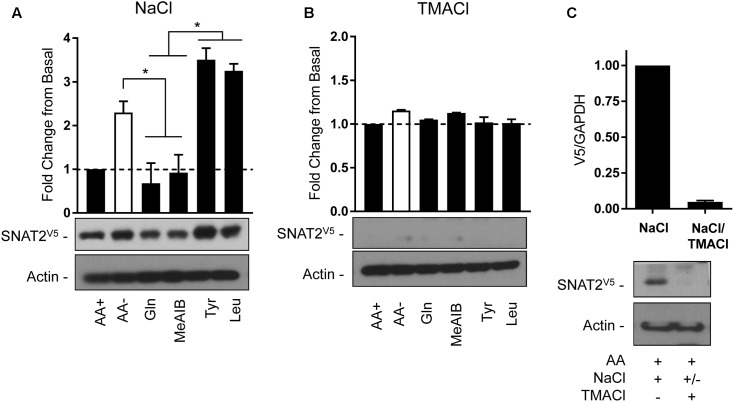
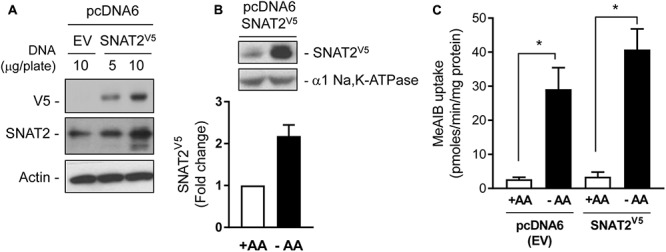
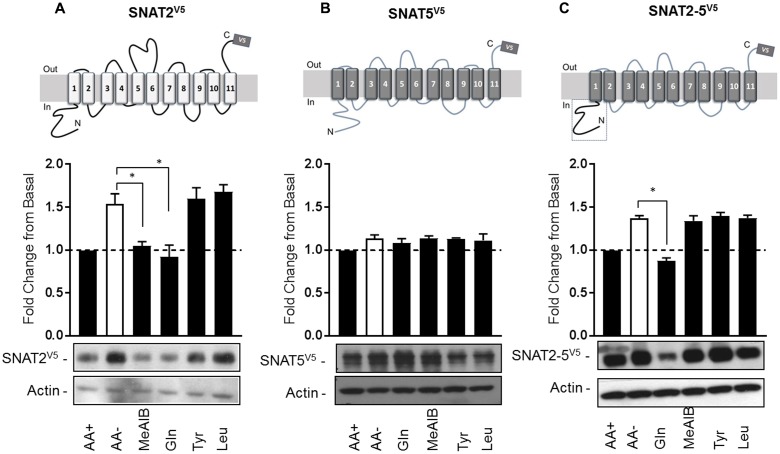
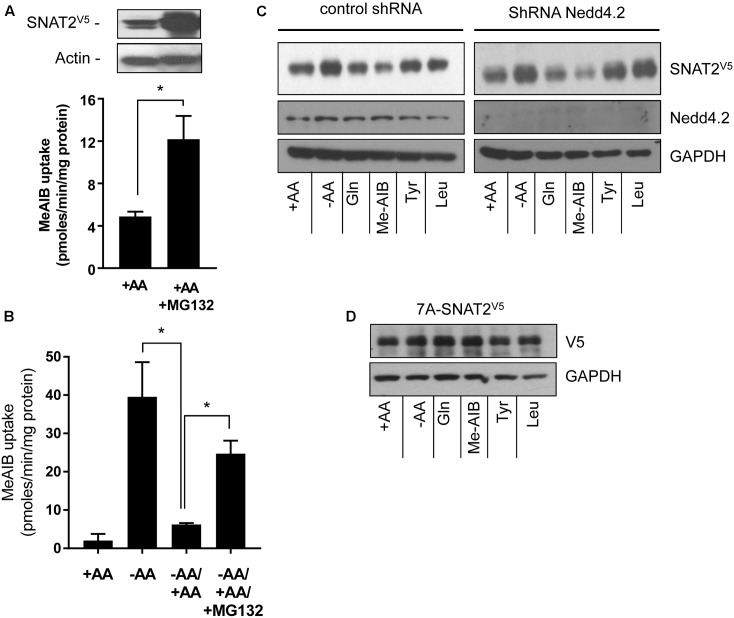
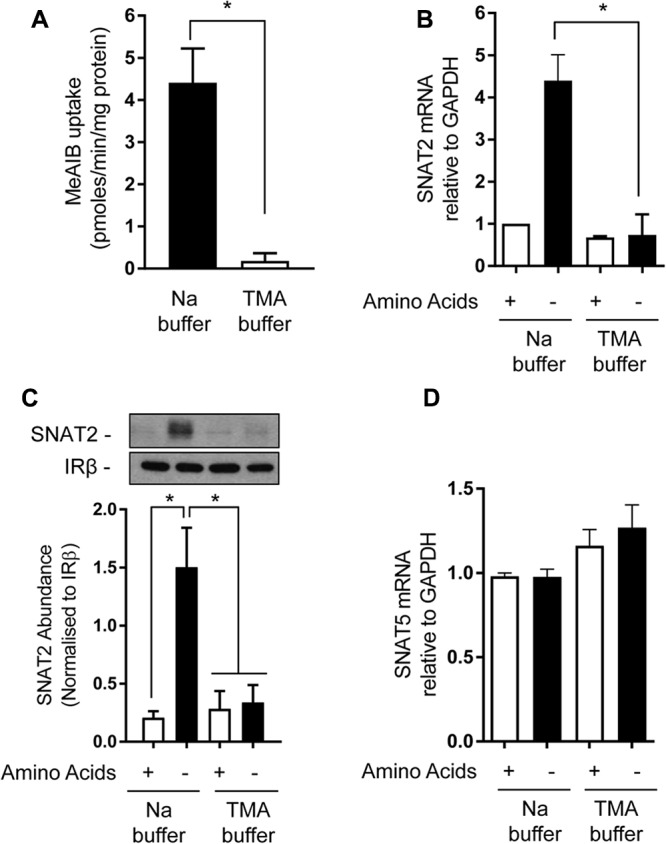
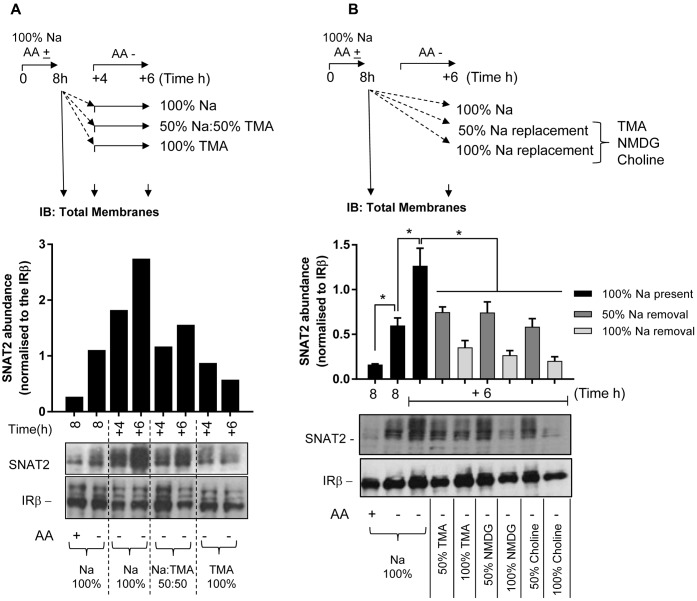
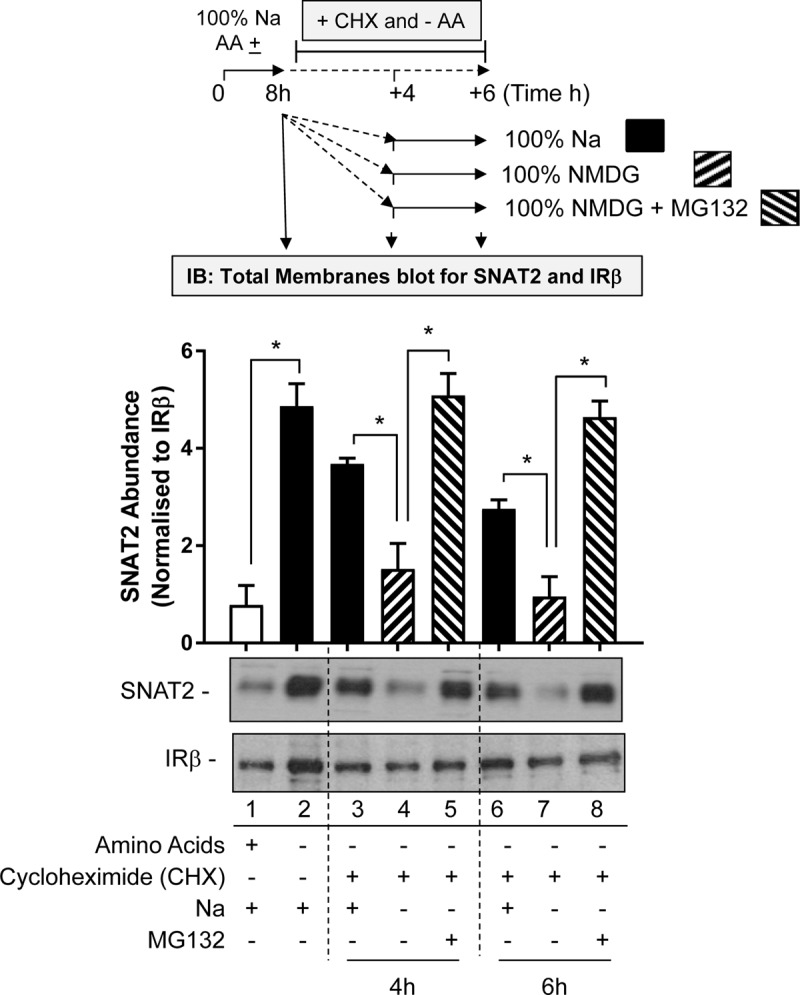
The SNAT2 (SLC38A2) System A amino acid transporter mediates Na-coupled cellular uptake of small neutral α-amino acids (AAs) and is extensively regulated in response to humoral and nutritional cues. Understanding the basis of such regulation is important given that AA uptake SNAT2 has been linked to activation of mTORC1; a major controller of many important cellular processes including, for example, mRNA translation, lipid synthesis, and autophagy and whose dysregulation has been implicated in the development of cancer and conditions such as obesity and type 2 diabetes. Extracellular AA withdrawal induces an adaptive upregulation of SNAT2 gene transcription and SNAT2 protein stability but, as yet, the sensing mechanism(s) that initiate this response remain poorly understood although interactions between SNAT2 and its substrates may play a vital role. Herein, we have explored how changes in substrate (AA and Na) availability impact upon the adaptive regulation of SNAT2 in HeLa cells. We show that while AA deprivation induces SNAT2 gene expression, this induction was not apparent if extracellular Na was removed during the AA withdrawal period. Furthermore, we show that the increase in SNAT2 protein stability associated with AA withdrawal is selectively repressed by provision of SNAT2 AA substrates (-methylaminoisobutyric acid and glutamine), but not non-substrates. This stabilization and substrate-induced repression were critically dependent upon the cytoplasmic N-terminal tail of SNAT2 (containing lysyl residues which are putative targets of the ubiquitin-proteasome system), because "grafting" this tail onto SNAT5, a related SLC38 family member that does not exhibit adaptive regulation, confers substrate-induced changes in stability of the SNAT2-5 chimeric transporter. In contrast, expression of SNAT2 in which the N-terminal lysyl residues were mutated to alanine rendered the transporter stable and insensitive to substrate-induced changes in protein stability. Intriguingly, SNAT2 protein stability was dramatically reduced in the absence of extracellular Na irrespective of whether substrate AAs were present or absent. Our findings indicate that the presence of extracellular Na (and potentially its binding to SNAT2) may be crucial for not only sensing SNAT2 AA occupancy and consequently for initiating the adaptive response under AA insufficient conditions, but for enabling substrate-induced changes in SNAT2 protein stability.
系统A氨基酸转运体SNAT2(溶质载体家族38成员2,SLC38A2)介导中性小α-氨基酸(AAs)的钠偶联细胞摄取,并受体液和营养信号广泛调控。鉴于AA通过SNAT2摄取与mTORC1的激活有关,而mTORC1是许多重要细胞过程(如mRNA翻译、脂质合成和自噬)的主要调控因子,其失调与癌症以及肥胖症和2型糖尿病等疾病的发生有关,因此了解这种调控的基础非常重要。细胞外AA撤除会诱导SNAT2基因转录和SNAT2蛋白稳定性的适应性上调,但迄今为止,引发这种反应的传感机制仍知之甚少,尽管SNAT2与其底物之间的相互作用可能起着至关重要的作用。在此,我们探讨了底物(AA和Na)可用性的变化如何影响HeLa细胞中SNAT2的适应性调控。我们发现,虽然AA剥夺会诱导SNAT2基因表达,但如果在AA撤除期间去除细胞外Na,这种诱导并不明显。此外,我们发现,与AA撤除相关的SNAT2蛋白稳定性增加会被提供SNAT2 AA底物(-甲基氨基异丁酸和谷氨酰胺)选择性抑制,但不会被非底物抑制。这种稳定性以及底物诱导的抑制作用关键取决于SNAT2的细胞质N末端尾巴(含有赖氨酸残基,这些残基是泛素-蛋白酶体系统的假定靶点),因为将这个尾巴“嫁接”到不表现出适应性调控的相关SLC38家族成员SNAT5上,会使SNAT2-5嵌合转运体的稳定性发生底物诱导的变化。相反,将N末端赖氨酸残基突变为丙氨酸的SNAT2表达使转运体稳定,且对底物诱导的蛋白稳定性变化不敏感。有趣的是,无论底物AAs是否存在,在没有细胞外Na的情况下,SNAT2蛋白稳定性都会显著降低。我们的研究结果表明,细胞外Na的存在(以及其可能与SNAT2的结合)不仅对于感知SNAT2的AA占据情况并因此在AA不足的条件下启动适应性反应至关重要,而且对于实现底物诱导的SNAT2蛋白稳定性变化也至关重要。