Mehta Paul, Kaye Wendy, Raymond Jaime, Wu Ruoming, Larson Theodore, Punjani Reshma, Heller Daniel, Cohen Jessica, Peters Tracy, Muravov Oleg, Horton Kevin
Division of Toxicology and Human Health Sciences, Agency for Toxic Substances and Disease Registry, CDC.
MMWR Morb Mortal Wkly Rep. 2018 Feb 23;67(7):216-218. doi: 10.15585/mmwr.mm6707a3.
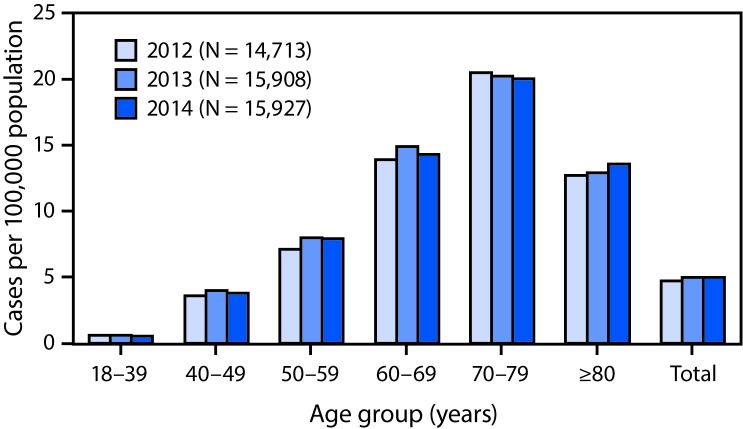
Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig's disease, is a progressive and fatal neuromuscular disease; the majority of ALS patients die within 2-5 years of receiving a diagnosis (1). Familial ALS, a hereditary form of the disease, accounts for 5%-10% of cases, whereas the remaining sporadic cases have no clearly defined etiology (1). ALS affects persons of all races and ethnicities; however, whites, males, non-Hispanics, persons aged >60 years, and those with a family history of ALS are more likely to develop the disease (1-3). No cure for ALS has yet been identified, and the lack of proven and effective therapeutic interventions is an ongoing challenge. Current treatments available do not cure ALS but have been shown to slow disease progression. Until recently, only one drug (riluzole) was approved to treat ALS; however, in 2017, the Food and Drug Administration approved a second drug, edaravone (4).
肌萎缩侧索硬化症(ALS),通常被称为卢伽雷氏病,是一种进行性的致命神经肌肉疾病;大多数ALS患者在确诊后2至5年内死亡(1)。家族性ALS是该疾病的一种遗传形式,占病例的5%至10%,而其余的散发性病例没有明确的病因(1)。ALS影响所有种族和族裔的人群;然而,白人、男性、非西班牙裔、60岁以上的人以及有ALS家族史的人更易患此病(1 - 3)。目前尚未找到治愈ALS的方法,缺乏经过验证的有效治疗干预措施仍是一个持续存在的挑战。现有的治疗方法无法治愈ALS,但已证明可减缓疾病进展。直到最近,只有一种药物(利鲁唑)被批准用于治疗ALS;然而,在2017年,美国食品药品监督管理局批准了第二种药物,依达拉奉(4)。