Shah Nidhi, Altschul Stephen F, Pop Mihai
1Department of Computer Science and Center for Bioinformatics and Computational Biology, University of Maryland, College Park, 20742 USA.
2Computational Biology Branch, NCBI, NLM, NIH, Bethesda, 20894 USA.
Algorithms Mol Biol. 2018 Mar 22;13:7. doi: 10.1186/s13015-018-0126-3. eCollection 2018.
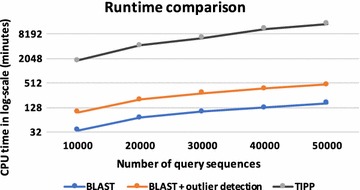
An important task in a metagenomic analysis is the assignment of taxonomic labels to sequences in a sample. Most widely used methods for taxonomy assignment compare a sequence in the sample to a database of known sequences. Many approaches use the best BLAST hit(s) to assign the taxonomic label. However, it is known that the best BLAST hit may not always correspond to the best taxonomic match. An alternative approach involves phylogenetic methods, which take into account alignments and a model of evolution in order to more accurately define the taxonomic origin of sequences. Similarity-search based methods typically run faster than phylogenetic methods and work well when the organisms in the sample are well represented in the database. In contrast, phylogenetic methods have the capability to identify new organisms in a sample but are computationally quite expensive.
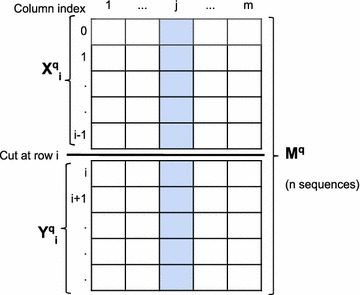
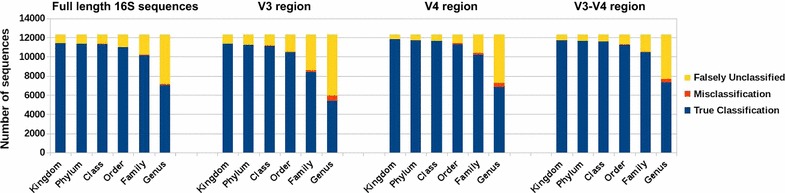
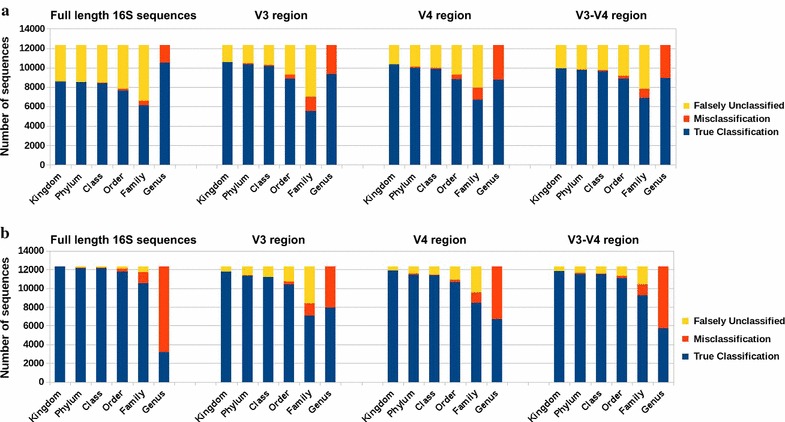
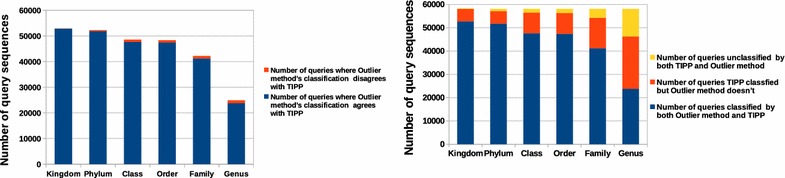
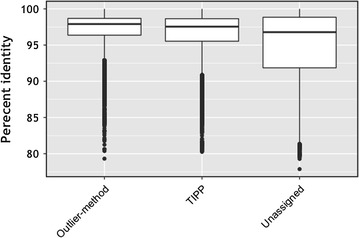
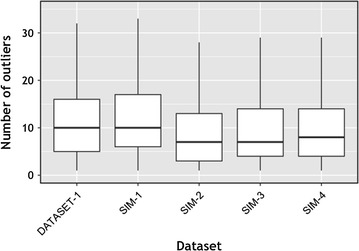
We propose a two-step approach for metagenomic taxon identification; i.e., use a rapid method that accurately classifies sequences using a reference database (this is a filtering step) and then use a more complex phylogenetic method for the sequences that were unclassified in the previous step. In this work, we explore whether and when using top BLAST hit(s) yields a correct taxonomic label. We develop a method to detect outliers among BLAST hits in order to separate the phylogenetically most closely related matches from matches to sequences from more distantly related organisms. We used modified BILD (Bayesian Integral Log-Odds) scores, a multiple-alignment scoring function, to define the outliers within a subset of top BLAST hits and assign taxonomic labels. We compared the accuracy of our method to the RDP classifier and show that our method yields fewer misclassifications while properly classifying organisms that are not present in the database. Finally, we evaluated the use of our method as a pre-processing step before more expensive phylogenetic analyses (in our case TIPP) in the context of real 16S rRNA datasets.
Our experiments make a good case for using a two-step approach for accurate taxonomic assignment. We show that our method can be used as a filtering step before using phylogenetic methods and provides a way to interpret BLAST results using more information than provided by E-values and bit-scores alone.
宏基因组分析中的一项重要任务是为样本中的序列分配分类标签。最广泛使用的分类学分配方法是将样本中的序列与已知序列数据库进行比较。许多方法使用最佳的BLAST比对结果来分配分类标签。然而,众所周知,最佳的BLAST比对结果可能并不总是对应于最佳的分类学匹配。另一种方法涉及系统发育方法,该方法考虑比对和进化模型,以便更准确地定义序列的分类学起源。基于相似性搜索的方法通常比系统发育方法运行得更快,并且当样本中的生物在数据库中有很好的代表性时效果良好。相比之下,系统发育方法有能力识别样本中的新生物,但计算成本相当高。
我们提出了一种用于宏基因组分类单元识别的两步法;即,使用一种快速方法,通过参考数据库准确地对序列进行分类(这是一个过滤步骤),然后对前一步中未分类的序列使用更复杂的系统发育方法。在这项工作中,我们探索使用最佳BLAST比对结果是否以及何时能产生正确的分类标签。我们开发了一种方法来检测BLAST比对结果中的异常值,以便将系统发育上最密切相关的匹配与来自更远缘相关生物的序列的匹配区分开来。我们使用修改后的BILD(贝叶斯积分对数似然)分数,一种多重比对评分函数,来定义最佳BLAST比对结果子集中的异常值并分配分类标签。我们将我们方法的准确性与RDP分类器进行了比较,结果表明我们的方法产生的错误分类更少,同时能正确分类数据库中不存在的生物。最后,我们在真实的16S rRNA数据集的背景下,评估了我们的方法作为在更昂贵的系统发育分析(在我们的案例中是TIPP)之前的预处理步骤的用途。
我们的实验有力地支持了使用两步法进行准确的分类学分配。我们表明,我们的方法可以在使用系统发育方法之前用作过滤步骤,并提供了一种比单独使用E值和比特分数更多信息来解释BLAST结果的方法。