Gao Xiang, Lin Huaiying, Revanna Kashi, Dong Qunfeng
Department of Public Health Sciences, Loyola University Chicago Health Sciences Division, Maywood, IL, 60153, USA.
Center for Biomedical Informatics, Loyola University Chicago Health Sciences Division, Maywood, IL, 60153, USA.
BMC Bioinformatics. 2017 May 10;18(1):247. doi: 10.1186/s12859-017-1670-4.
Species-level classification for 16S rRNA gene sequences remains a serious challenge for microbiome researchers, because existing taxonomic classification tools for 16S rRNA gene sequences either do not provide species-level classification, or their classification results are unreliable. The unreliable results are due to the limitations in the existing methods which either lack solid probabilistic-based criteria to evaluate the confidence of their taxonomic assignments, or use nucleotide k-mer frequency as the proxy for sequence similarity measurement.
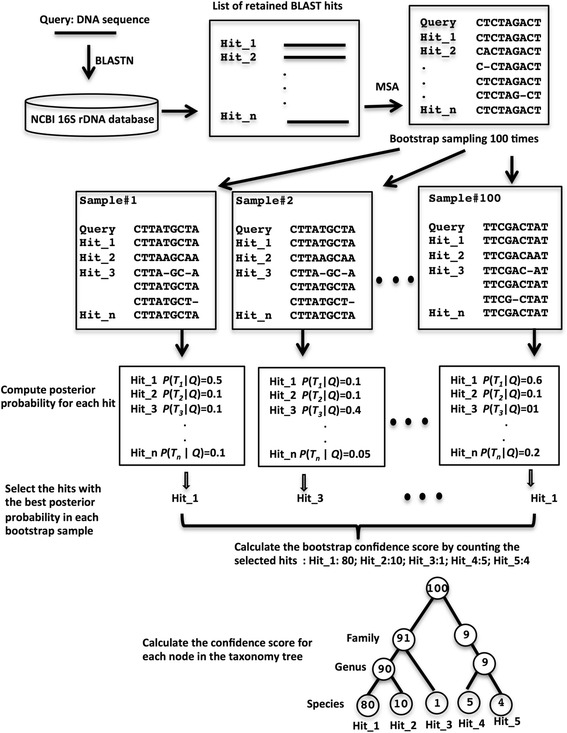
We have developed a method that shows significantly improved species-level classification results over existing methods. Our method calculates true sequence similarity between query sequences and database hits using pairwise sequence alignment. Taxonomic classifications are assigned from the species to the phylum levels based on the lowest common ancestors of multiple database hits for each query sequence, and further classification reliabilities are evaluated by bootstrap confidence scores. The novelty of our method is that the contribution of each database hit to the taxonomic assignment of the query sequence is weighted by a Bayesian posterior probability based upon the degree of sequence similarity of the database hit to the query sequence. Our method does not need any training datasets specific for different taxonomic groups. Instead only a reference database is required for aligning to the query sequences, making our method easily applicable for different regions of the 16S rRNA gene or other phylogenetic marker genes.
Reliable species-level classification for 16S rRNA or other phylogenetic marker genes is critical for microbiome research. Our software shows significantly higher classification accuracy than the existing tools and we provide probabilistic-based confidence scores to evaluate the reliability of our taxonomic classification assignments based on multiple database matches to query sequences. Despite its higher computational costs, our method is still suitable for analyzing large-scale microbiome datasets for practical purposes. Furthermore, our method can be applied for taxonomic classification of any phylogenetic marker gene sequences. Our software, called BLCA, is freely available at https://github.com/qunfengdong/BLCA .
对于微生物组研究人员而言,16S rRNA基因序列的物种水平分类仍然是一项严峻挑战,因为现有的16S rRNA基因序列分类工具要么不提供物种水平分类,要么其分类结果不可靠。结果不可靠是由于现有方法存在局限性,这些方法要么缺乏基于可靠概率的标准来评估其分类归属的可信度,要么使用核苷酸k-mer频率作为序列相似性测量的替代指标。
我们开发了一种方法,与现有方法相比,该方法在物种水平分类结果上有显著改进。我们的方法使用成对序列比对来计算查询序列与数据库匹配序列之间的真实序列相似性。基于每个查询序列的多个数据库匹配的最低共同祖先,从物种到门水平进行分类归属,并通过自展置信度得分评估进一步的分类可靠性。我们方法的新颖之处在于,每个数据库匹配对查询序列分类归属的贡献通过基于数据库匹配与查询序列的序列相似性程度的贝叶斯后验概率进行加权。我们的方法不需要针对不同分类组的任何训练数据集。相反,只需要一个参考数据库来与查询序列进行比对,这使得我们的方法易于应用于16S rRNA基因的不同区域或其他系统发育标记基因。
对16S rRNA或其他系统发育标记基因进行可靠的物种水平分类对于微生物组研究至关重要。我们的软件显示出比现有工具显著更高的分类准确性,并且我们基于多个数据库与查询序列的匹配提供基于概率的置信度得分,以评估我们分类归属的可靠性。尽管计算成本较高,但我们的方法仍然适用于实际分析大规模微生物组数据集。此外,我们的方法可应用于任何系统发育标记基因序列的分类。我们的软件名为BLCA,可在https://github.com/qunfengdong/BLCA上免费获取。