Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain.
Universitat Pompeu Fabra (UPF), Barcelona, Spain.
Nucleic Acids Res. 2018 May 4;46(8):3852-3863. doi: 10.1093/nar/gky228.
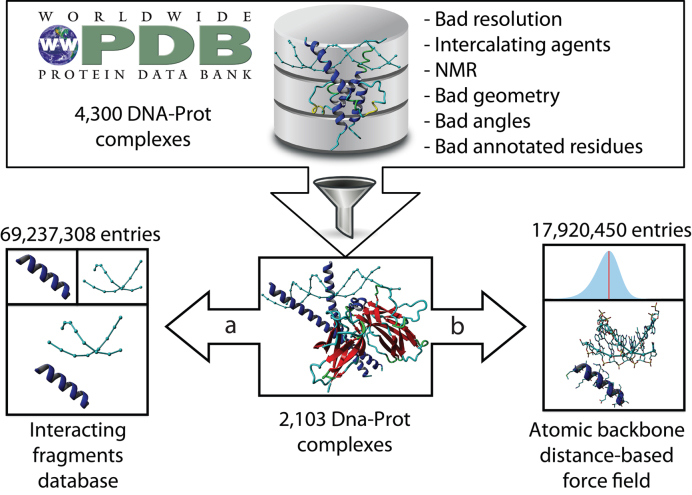
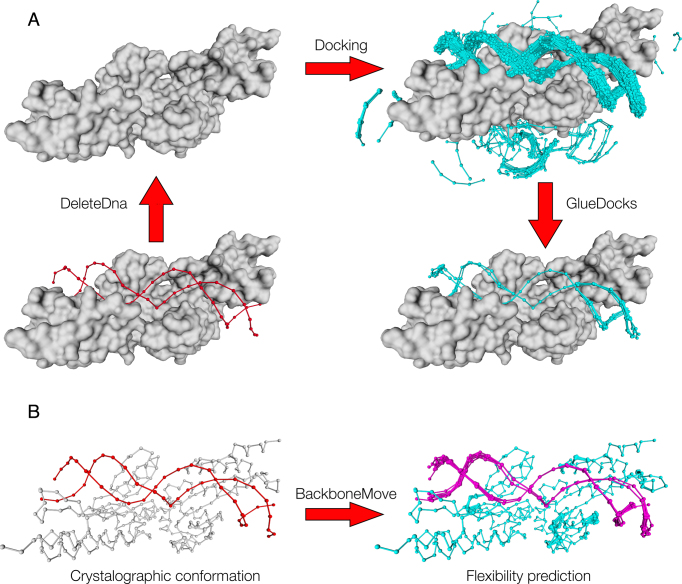
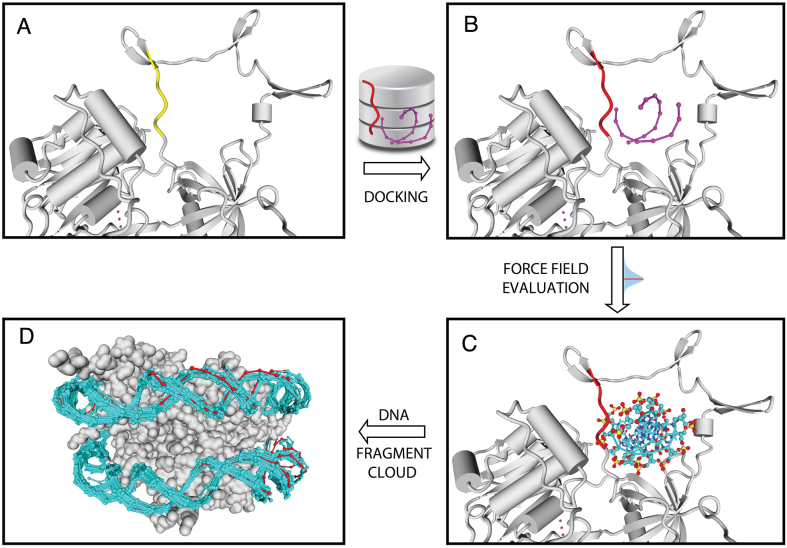
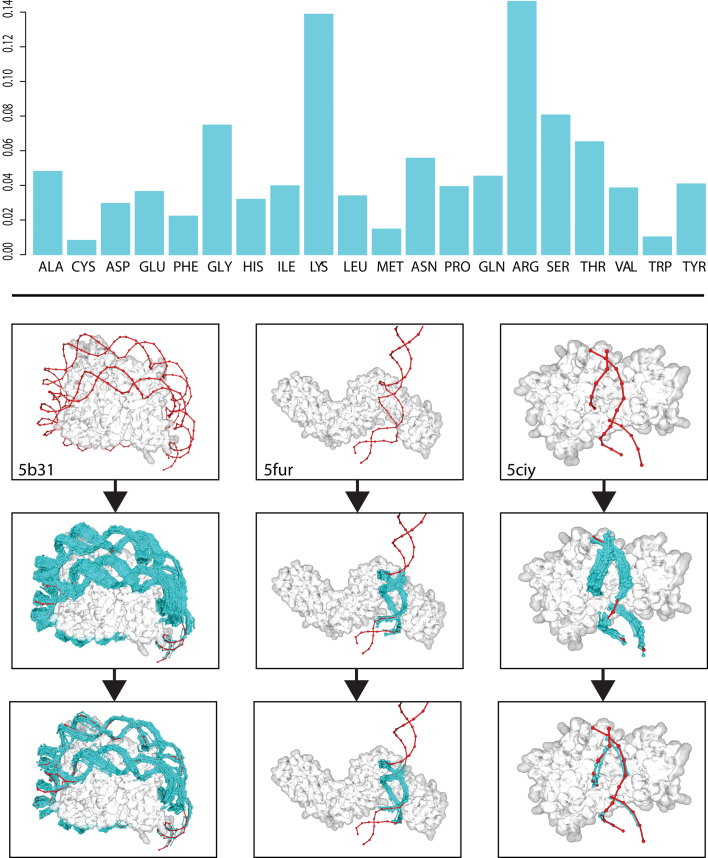
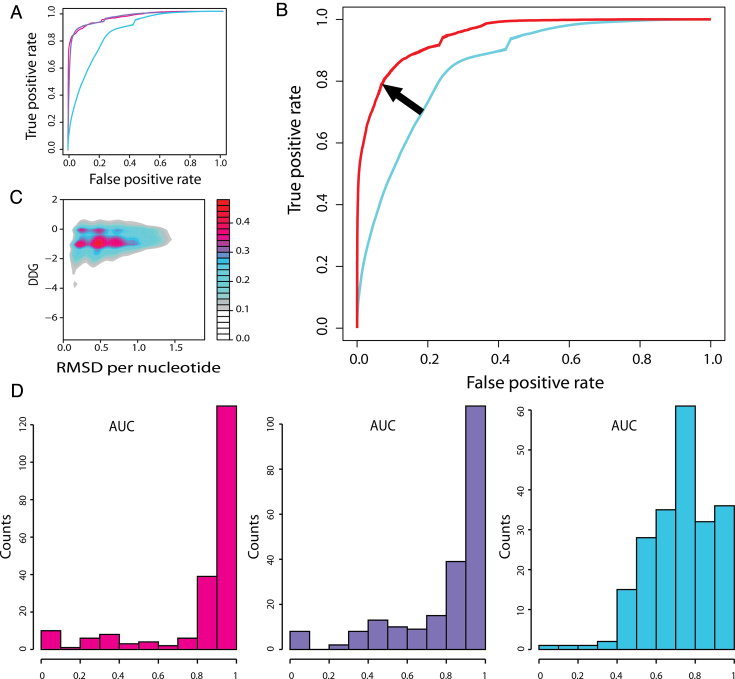
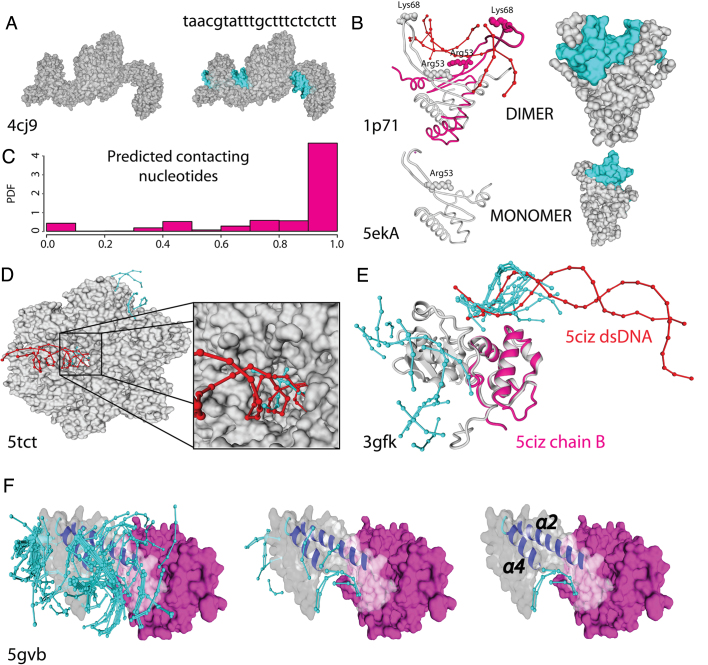
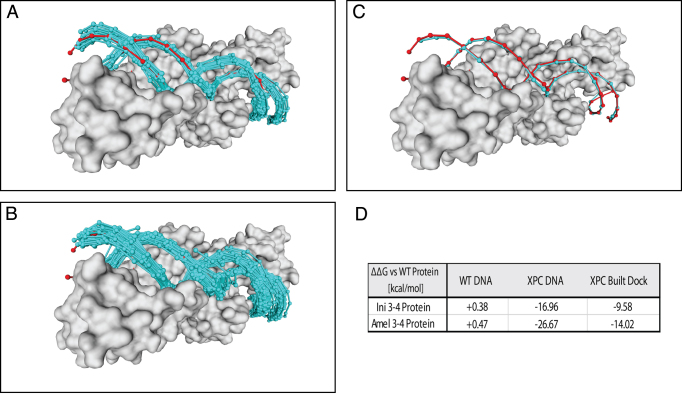
The speed at which new genomes are being sequenced highlights the need for genome-wide methods capable of predicting protein-DNA interactions. Here, we present PADA1, a generic algorithm that accurately models structural complexes and predicts the DNA-binding regions of resolved protein structures. PADA1 relies on a library of protein and double-stranded DNA fragment pairs obtained from a training set of 2103 DNA-protein complexes. It includes a fast statistical force field computed from atom-atom distances, to evaluate and filter the 3D docking models. Using published benchmark validation sets and 212 DNA-protein structures published after 2016 we predicted the DNA-binding regions with an RMSD of <1.8 Å per residue in >95% of the cases. We show that the quality of the docked templates is compatible with FoldX protein design tool suite to identify the crystallized DNA molecule sequence as the most energetically favorable in 80% of the cases. We highlighted the biological potential of PADA1 by reconstituting DNA and protein conformational changes upon protein mutagenesis of a meganuclease and its variants, and by predicting DNA-binding regions and nucleotide sequences in proteins crystallized without DNA. These results opens up new perspectives for the engineering of DNA-protein interfaces.
新基因组测序的速度凸显了对能够预测蛋白质-DNA 相互作用的全基因组方法的需求。在这里,我们提出了 PADA1,这是一种通用算法,能够准确地模拟结构复合物,并预测已解析蛋白质结构的 DNA 结合区域。PADA1 依赖于从 2103 个 DNA-蛋白质复合物的训练集中获得的蛋白质和双链 DNA 片段对的库。它包括一个从原子间距离计算得出的快速统计力场,用于评估和筛选 3D 对接模型。使用已发表的基准验证集和 2016 年后发表的 212 个 DNA-蛋白质结构,我们预测 DNA 结合区域的 RMSD 小于 1.8 Å/残基,在>95%的情况下。我们表明,对接模板的质量与 FoldX 蛋白质设计工具套件兼容,可识别结晶 DNA 分子序列作为 80%情况下最具能量优势的序列。我们通过重新构建在核酸酶及其变体的蛋白质突变后 DNA 和蛋白质构象变化,以及预测没有 DNA 的蛋白质结晶中的 DNA 结合区域和核苷酸序列,突出了 PADA1 的生物学潜力。这些结果为 DNA-蛋白质界面的工程设计开辟了新的前景。