Price E M, Robinson Wendy P
BC Children's Hospital Research Institute, Vancouver, BC, Canada.
Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada.
Front Genet. 2018 Mar 16;9:83. doi: 10.3389/fgene.2018.00083. eCollection 2018.
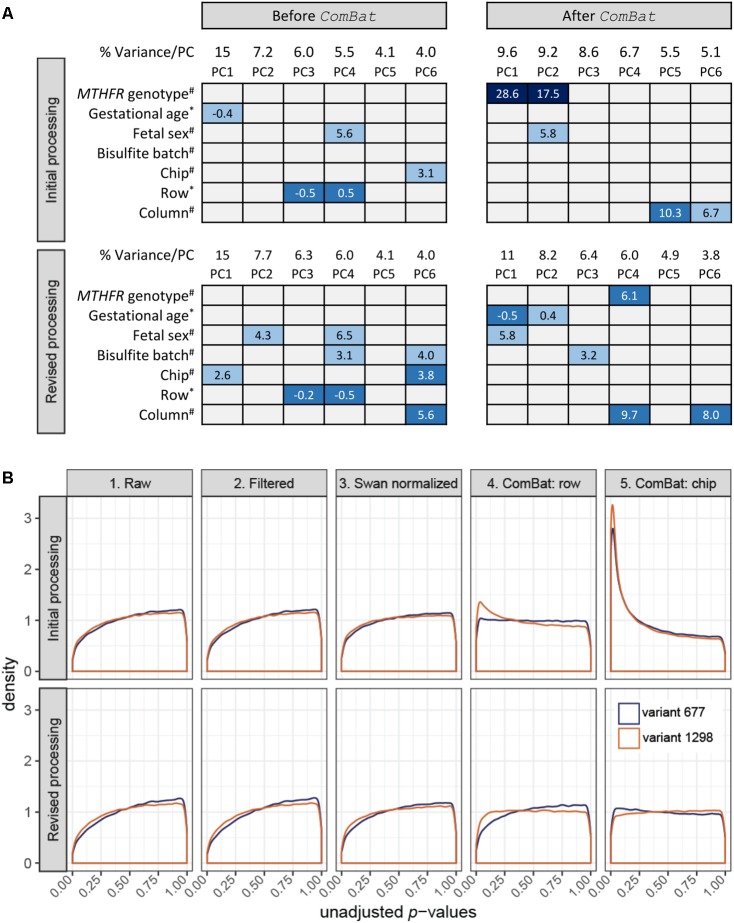
It is well-known, but frequently overlooked, that low- and high-throughput molecular data may contain batch effects, i.e., systematic technical variation. Confounding of experimental batches with the variable(s) of interest is especially concerning, as a batch effect may then be interpreted as a biologically significant finding. An integral step toward reducing false discovery in molecular data analysis includes inspection for batch effects and accounting for this signal if present. In a 30-sample pilot Illumina Infinium HumanMethylation450 (450k array) experiment, we identified two sources of batch effects: row and chip. Here, we demonstrate two approaches taken to process the 450k data in which an R function, , was applied to adjust for the non-biological signal. In the "initial analysis," the application of ComBat to an unbalanced study design resulted in 9,612 and 19,214 significant (FDR < 0.05) DNA methylation differences, despite none present prior to correction. Suspicious of this dramatic change, a "revised processing" included changes to our analysis as well as a greater number of samples, and successfully reduced batch effects without introducing false signal. Our work supports conclusions made by an article previously published in this journal: though the ultimate antidote to batch effects is thoughtful study design, every DNA methylation microarray analysis should inspect, assess and, if necessary, account for batch effects. The analysis experience presented here can serve as a reminder to the broader community to establish research questions , ensure that they match with study design and encourage communication between technicians and analysts.
众所周知,但经常被忽视的是,低通量和高通量分子数据可能包含批次效应,即系统性技术变异。实验批次与感兴趣的变量之间的混淆尤其令人担忧,因为批次效应可能会被解释为具有生物学意义的发现。减少分子数据分析中错误发现的一个不可或缺的步骤包括检查批次效应,并在存在这种信号时对其进行处理。在一项包含30个样本的Illumina Infinium HumanMethylation450(450k阵列)先导实验中,我们识别出了两种批次效应来源:行和芯片。在这里,我们展示了处理450k数据所采用的两种方法,其中应用了一个R函数来调整非生物学信号。在“初始分析”中,将ComBat应用于不平衡的研究设计导致了9612个和19214个显著(FDR < 0.05)的DNA甲基化差异,尽管在校正之前不存在差异。由于怀疑这种巨大变化,“修订处理”包括对我们的分析进行更改以及增加样本数量,并成功减少了批次效应而未引入错误信号。我们的工作支持了该期刊之前发表的一篇文章所得出的结论:尽管消除批次效应的最终方法是精心设计研究,但每次DNA甲基化微阵列分析都应检查、评估并在必要时处理批次效应。这里呈现的分析经验可以提醒更广泛的群体确立研究问题,确保它们与研究设计相匹配,并鼓励技术人员和分析人员之间的沟通。