Department of Medical Oncology, Virgen de la Salud Hospital, Toledo, Spain.
Institute of Biopathology and Regenerative Medicine (IBIMER), Center of Biomedical Research (CIBM), University of Granada, Granada, Spain.
PLoS One. 2018 Apr 4;13(4):e0194844. doi: 10.1371/journal.pone.0194844. eCollection 2018.
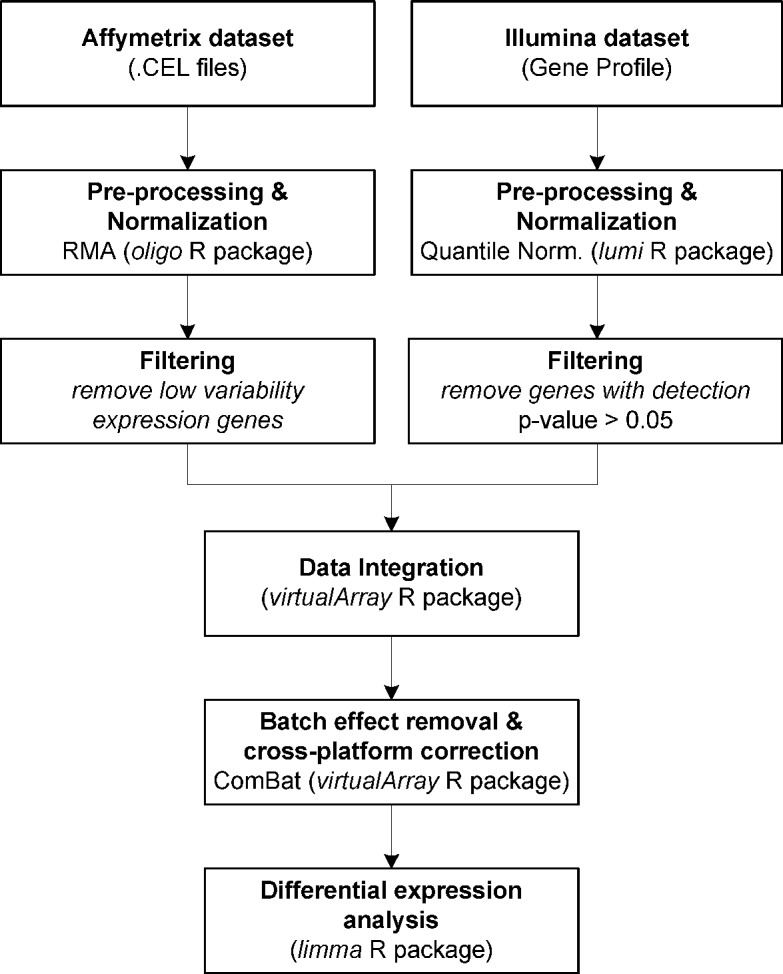
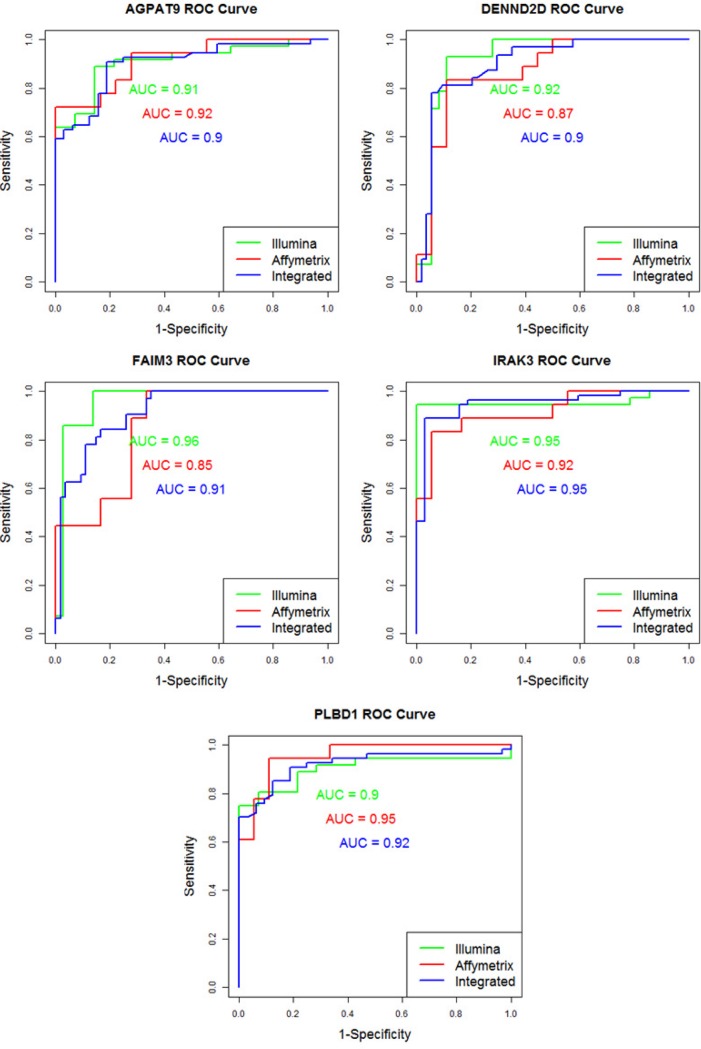
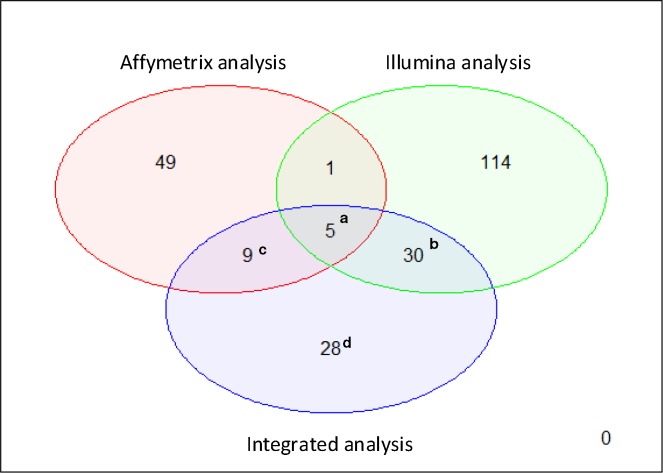
Applying differentially expressed genes (DEGs) to identify feasible biomarkers in diseases can be a hard task when working with heterogeneous datasets. Expression data are strongly influenced by technology, sample preparation processes, and/or labeling methods. The proliferation of different microarray platforms for measuring gene expression increases the need to develop models able to compare their results, especially when different technologies can lead to signal values that vary greatly. Integrative meta-analysis can significantly improve the reliability and robustness of DEG detection. The objective of this work was to develop an integrative approach for identifying potential cancer biomarkers by integrating gene expression data from two different platforms. Pancreatic ductal adenocarcinoma (PDAC), where there is an urgent need to find new biomarkers due its late diagnosis, is an ideal candidate for testing this technology. Expression data from two different datasets, namely Affymetrix and Illumina (18 and 36 PDAC patients, respectively), as well as from 18 healthy controls, was used for this study. A meta-analysis based on an empirical Bayesian methodology (ComBat) was then proposed to integrate these datasets. DEGs were finally identified from the integrated data by using the statistical programming language R. After our integrative meta-analysis, 5 genes were commonly identified within the individual analyses of the independent datasets. Also, 28 novel genes that were not reported by the individual analyses ('gained' genes) were also discovered. Several of these gained genes have been already related to other gastroenterological tumors. The proposed integrative meta-analysis has revealed novel DEGs that may play an important role in PDAC and could be potential biomarkers for diagnosing the disease.
应用差异表达基因 (DEGs) 来识别疾病中的可行生物标志物,当处理异质数据集时,这可能是一项艰巨的任务。表达数据受技术、样本制备过程和/或标记方法的强烈影响。用于测量基因表达的不同微阵列平台的激增增加了开发能够比较其结果的模型的需求,尤其是当不同的技术可能导致信号值差异很大时。综合荟萃分析可以显著提高 DEG 检测的可靠性和稳健性。本工作的目的是开发一种综合方法,通过整合来自两个不同平台的基因表达数据来识别潜在的癌症生物标志物。胰腺导管腺癌 (PDAC) 是一种理想的候选者,因为它需要找到新的生物标志物,因为其诊断较晚。本研究使用了来自两个不同数据集的基因表达数据,即 Affymetrix 和 Illumina(分别有 18 和 36 名 PDAC 患者和 18 名健康对照者)。然后,提出了一种基于经验贝叶斯方法(ComBat)的荟萃分析来整合这些数据集。最后,使用统计编程语言 R 从整合数据中识别 DEGs。经过我们的综合荟萃分析,在独立数据集的个体分析中共同鉴定出 5 个基因。此外,还发现了 28 个个体分析未报道的新基因(“获得”基因)。这些获得的基因中的几个已经与其他胃肠肿瘤有关。所提出的综合荟萃分析揭示了可能在 PDAC 中发挥重要作用并可能成为诊断该疾病的潜在生物标志物的新的 DEGs。