Department of Sociology, Princeton University, Princeton, NJ, United States of America.
Graduate School of Education, Stanford University, Stanford, CA, United States of America.
PLoS One. 2018 Apr 4;13(4):e0194541. doi: 10.1371/journal.pone.0194541. eCollection 2018.
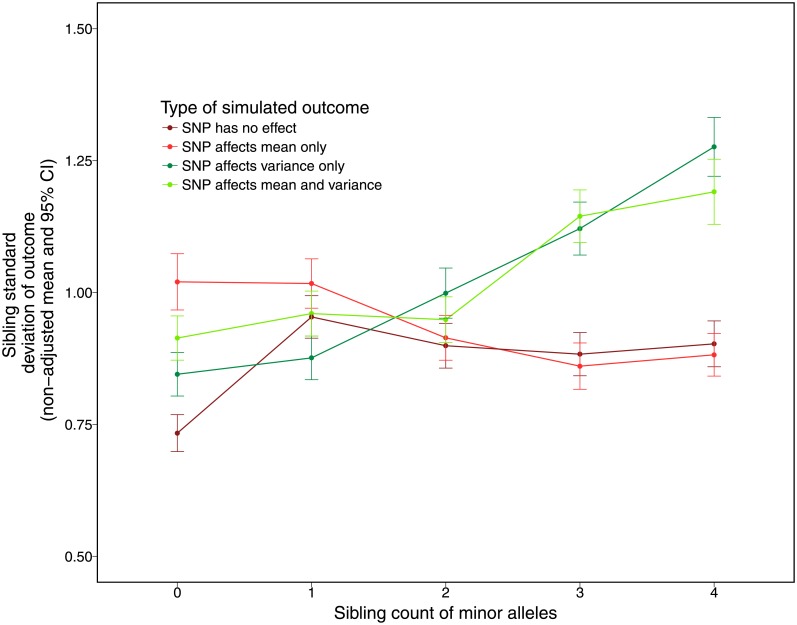
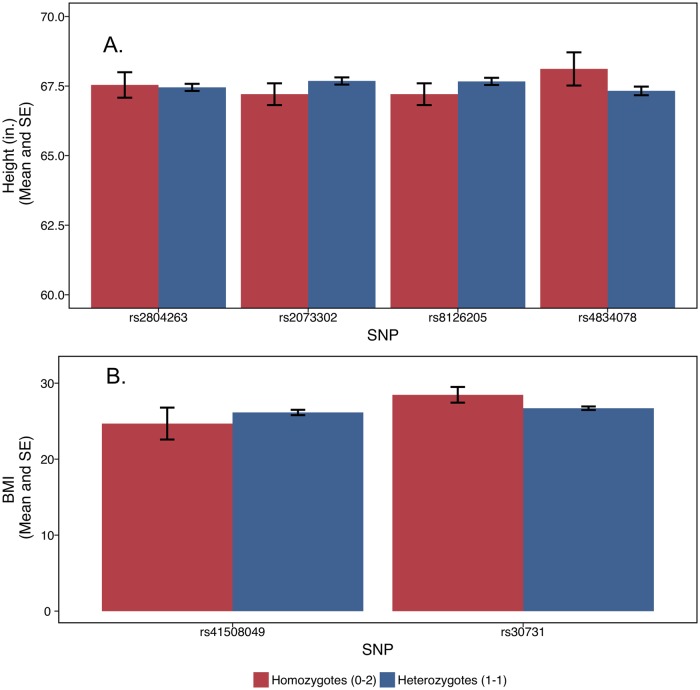
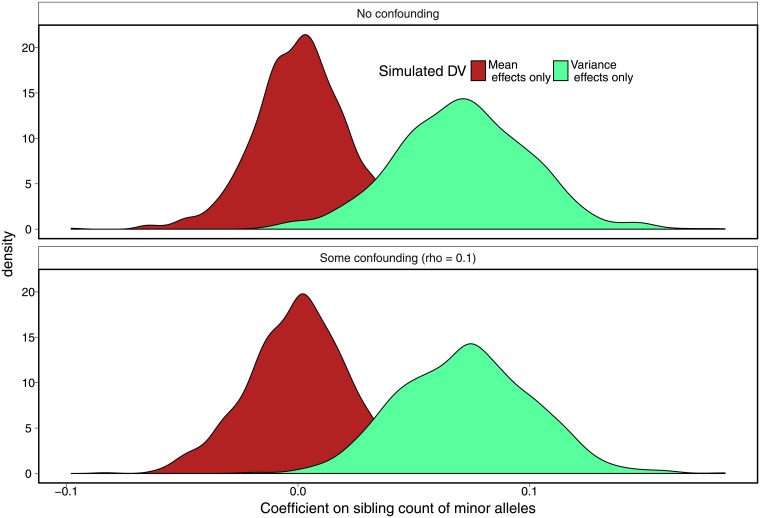
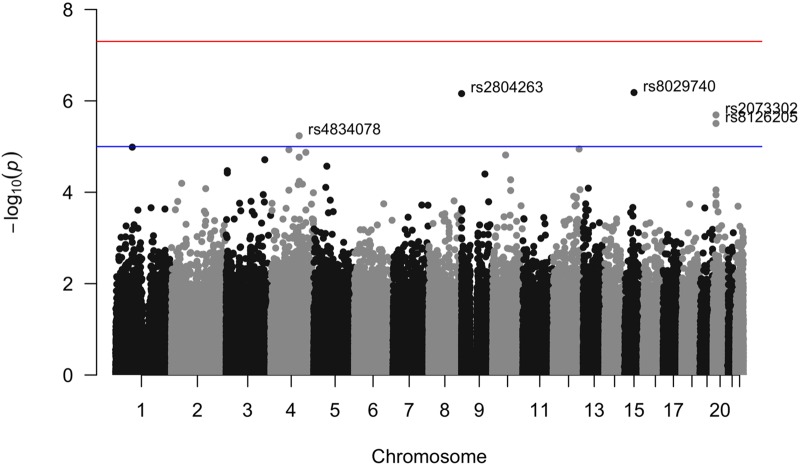
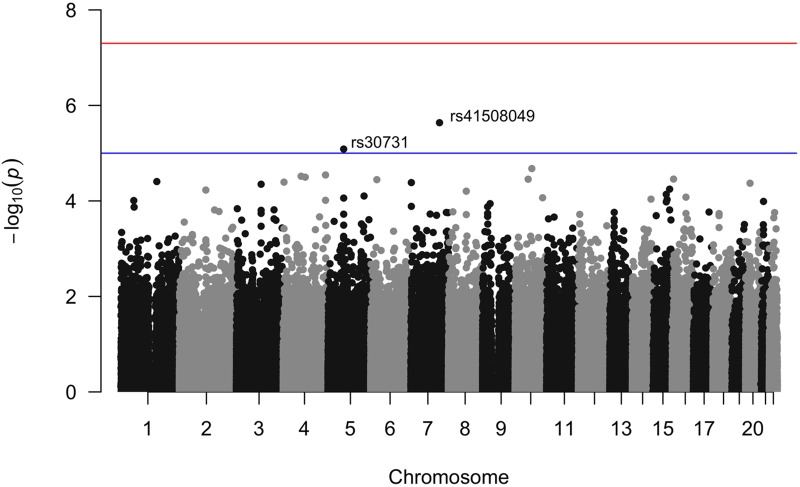
The propensity of a trait to vary within a population may have evolutionary, ecological, or clinical significance. In the present study we deploy sibling models to offer a novel and unbiased way to ascertain loci associated with the extent to which phenotypes vary (variance-controlling quantitative trait loci, or vQTLs). Previous methods for vQTL-mapping either exclude genetically related individuals or treat genetic relatedness among individuals as a complicating factor addressed by adjusting estimates for non-independence in phenotypes. The present method uses genetic relatedness as a tool to obtain unbiased estimates of variance effects rather than as a nuisance. The family-based approach, which utilizes random variation between siblings in minor allele counts at a locus, also allows controls for parental genotype, mean effects, and non-linear (dominance) effects that may spuriously appear to generate variation. Simulations show that the approach performs equally well as two existing methods (squared Z-score and DGLM) in controlling type I error rates when there is no unobserved confounding, and performs significantly better than these methods in the presence of small degrees of confounding. Using height and BMI as empirical applications, we investigate SNPs that alter within-family variation in height and BMI, as well as pathways that appear to be enriched. One significant SNP for BMI variability, in the MAST4 gene, replicated. Pathway analysis revealed one gene set, encoding members of several signaling pathways related to gap junction function, which appears significantly enriched for associations with within-family height variation in both datasets (while not enriched in analysis of mean levels). We recommend approximating laboratory random assignment of genotype using family data and more careful attention to the possible conflation of mean and variance effects.
性状在群体中变化的倾向可能具有进化、生态或临床意义。在本研究中,我们使用同胞模型提供了一种新颖且无偏的方法,以确定与表型变化程度相关的基因座(控制表型方差的数量性状基因座,或 vQTLs)。以前用于 vQTL 作图的方法要么排除遗传相关的个体,要么将个体之间的遗传相关性视为通过调整表型非独立性的估计来解决的复杂因素。本方法将遗传相关性用作获得方差效应无偏估计的工具,而不是作为干扰因素。基于家庭的方法利用了在一个基因座上的次要等位基因计数的兄弟姐妹之间的随机变异,还可以控制父母基因型、均值效应和非线性(显性)效应,这些效应可能会错误地产生变异。模拟表明,在不存在未观察到的混杂时,该方法在控制 I 型错误率方面与两种现有方法(平方 Z 分数和 DGLM)表现相当,并且在存在较小程度混杂时表现明显优于这些方法。使用身高和 BMI 作为实证应用,我们研究了改变身高和 BMI 家族内变异性的 SNP,以及似乎丰富的途径。一个与 BMI 变异性相关的 MAST4 基因中的显著 SNP 得到了复制。通路分析显示,一个基因集编码与缝隙连接功能相关的几个信号通路的成员,在两个数据集的家族内身高变化中都明显富集(而在均值水平分析中不富集)。我们建议使用家族数据近似模拟实验室随机分配基因型,并更加小心地注意均值和方差效应可能的混淆。