Yeh Yi-Chun, Needham David M, Sieradzki Ella T, Fuhrman Jed A
Department of Biological Sciences, University of Southern California, Los Angeles, California, USA.
mSystems. 2018 Apr 3;3(3). doi: 10.1128/mSystems.00023-18. eCollection 2018 May-Jun.
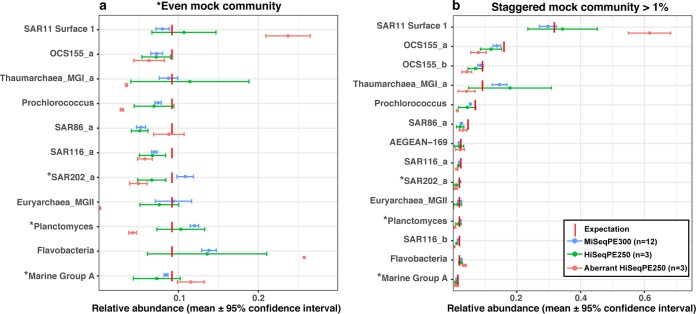
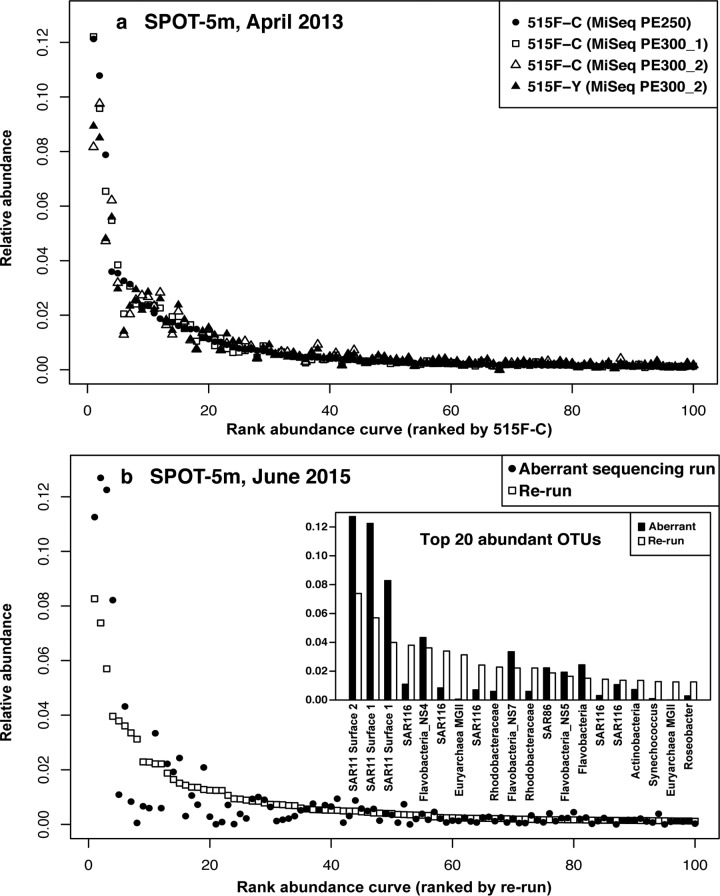
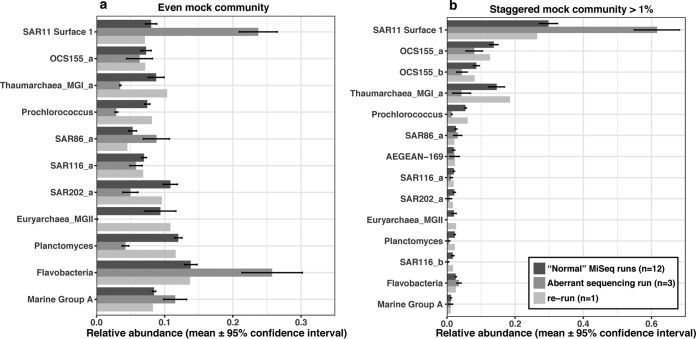
Mock communities have been used in microbiome method development to help estimate biases introduced in PCR amplification and sequencing and to optimize pipeline outputs. Nevertheless, the strong value of routine mock community analysis beyond initial method development is rarely, if ever, considered. Here we report that our routine use of mock communities as internal standards allowed us to discover highly aberrant and strong biases in the relative proportions of multiple taxa in a single Illumina HiSeqPE250 run. In this run, an important archaeal taxon virtually disappeared from all samples, and other mock community taxa showed >2-fold high or low abundance, whereas a rerun of those identical amplicons (from the same reaction tubes) on a different date yielded "normal" results. Although obvious from the strange mock community results, we could have easily missed the problem had we not used the mock communities because of natural variation of microbiomes at our site. The "normal" results were validated over four MiSeqPE300 runs and three HiSeqPE250 runs, and run-to-run variation was usually low. While validating these "normal" results, we also discovered that some mock microbial taxa had relatively modest, but consistent, differences between sequencing platforms. We strongly advise the use of mock communities in every sequencing run to distinguish potentially serious aberrations from natural variations. The mock communities should have more than just a few members and ideally at least partly represent the samples being analyzed to detect problems that show up only in some taxa and also to help validate clustering. Despite the routine use of standards and blanks in virtually all chemical or physical assays and most biological studies (a kind of "control"), microbiome analysis has traditionally lacked such standards. Here we show that unexpected problems of unknown origin can occur in such sequencing runs and yield completely incorrect results that would not necessarily be detected without the use of standards. Assuming that the microbiome sequencing analysis works properly every time risks serious errors that can be detected by the use of mock communities.
在微生物组方法开发中,已使用模拟群落来帮助评估PCR扩增和测序中引入的偏差,并优化流程输出。然而,除了最初的方法开发之外,常规模拟群落分析的强大价值很少(如果有的话)被考虑。在这里,我们报告称,我们将模拟群落作为内部标准的常规使用,使我们能够在一次Illumina HiSeqPE250运行中发现多个分类群相对比例中高度异常和强烈的偏差。在这次运行中,一个重要的古菌分类群实际上从所有样本中消失了,其他模拟群落分类群的丰度显示出高于或低于两倍的差异,而在不同日期对相同的扩增子(来自相同反应管)进行的重新运行产生了“正常”结果。尽管从奇怪的模拟群落结果中很明显,但如果我们没有使用模拟群落,由于我们现场微生物组的自然变异,我们很容易忽略这个问题。“正常”结果在四次MiSeqPE300运行和三次HiSeqPE250运行中得到了验证,并且运行间的变异通常较低。在验证这些“正常”结果的同时,我们还发现一些模拟微生物分类群在测序平台之间存在相对较小但一致的差异。我们强烈建议在每次测序运行中使用模拟群落,以区分潜在的严重偏差和自然变异。模拟群落应该不止有几个成员,理想情况下至少部分代表被分析的样本,以检测仅在某些分类群中出现的问题,并有助于验证聚类。尽管在几乎所有化学或物理分析以及大多数生物学研究中(一种“对照”)都常规使用标准品和空白对照,但微生物组分析传统上缺乏此类标准。在这里,我们表明在这样的测序运行中可能会出现来源不明的意外问题,并产生完全错误的结果,如果不使用标准品,这些结果不一定会被检测到。假设微生物组测序分析每次都能正常工作存在严重错误的风险,而使用模拟群落可以检测到这些错误。